
星形孢菌素生物合成途径的研究进展
2011-01-24 02:39赵雪尔刘骏陈敏
中国医药生物技术 2011年4期
赵雪尔,刘骏,陈敏
星形孢菌素(Staurosporine,STA)是从 Streptomyces Staurosporeus AM-2282 分离得到的一种生物碱。X 射线晶体结构分析发现它由一个吲哚咔唑核心和通过双 C-N 键连接的氨基己糖构成[1]。1986 年,人们发现它是一种对蛋白激酶 C(protein kinase C,PKC)非常有效的拮抗剂,因此该化合物被广泛用于各类细胞 PKC 在信号传导过程中所起作用的研究。STA 是目前已知的最有效的蛋白激酶抑制剂之一(IC50= 1 ~ 20 nmol/L)[2],由于其结构合成的复杂性和重要的生理活性,引起了众多有机合成化学家的兴趣。1995 年,Link 等[3]对它的合成全过程进行了描述。许多生物学家也对星形孢菌素的生物合成功能途径及改造进行了相关研究。本文将对星形孢菌素的来源,生物途径及改造工作进行综述。
1 星形孢菌素的生物合成基因簇
由于星形孢菌素的重要作用,人们对其生物合成途径展开研究。通过同位素标记 STA 产生菌内的前体化合物[4],发现吲哚咔唑核心来源于色氨酸,糖残基来源于葡萄糖,氨基糖连接后修饰的 O- 和 N-甲基的碳原子和质子直接来源于蛋氨酸[5]。
研究表明,STA 的生物合成基因簇由 15 个开放阅读框(open reading frame,ORF)组成,总长 22 kb[6-7],基因结构如图 1 所示。

图 1 星形孢菌素基因簇结构
星形孢菌素的生物合成路径如图 2 所示。中间产物CPA(chromopyrrolic acid)的形成需要 StaO 和 StaP 的催化。在 StaO 作用下,L-色氨酸氧化脱氨基启动合成反应发生,形成吲哚-3-丙酮酸(IPA)的亚胺形式。亚铁血红素蛋白四聚体 StaD 在 NH4+存在的条件下,催化两分子的 IPA产生 CPA[8]。StaD 家族的蛋白质由将近 1000 个氨基酸组成,氨基酸序列推导时发现它们没有明显保守区,这表明StaD 家族是血红素蛋白中新的一类,具有不同的结构和功能[9]。开放式的 CPA 必须转化成封闭式的吲哚咔唑糖苷配基,这就需要一对酶——StaP/StaC 催化形成吲哚咔唑核心[10-12],即 K252c。随后通过糖基化形成 STA。糖基化在STA 合成中十分重要,因为吲哚咔唑发挥生物活性需要糖基参与。糖残基通过糖基转移酶 StaG 连接氨基糖的 C-1’和 K252c 的 N-13,产生新的吲哚生物碱 holyrine A,StaN P450 连接 C-5’ 和 N-12 建立第二个 C-N 键,产生 O-去甲基-N-去甲基 STA,随后 StaMA 和 StaMB 甲基转移酶连续作用于这个中间产物,最终形成 STA[13]。各基因在STA 基因簇中的作用如表 1。
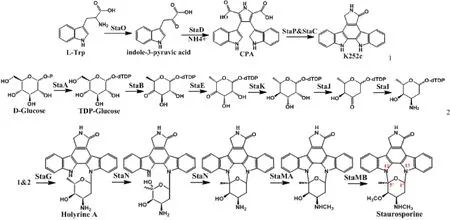
图 2 星形孢菌素的生物合成路径
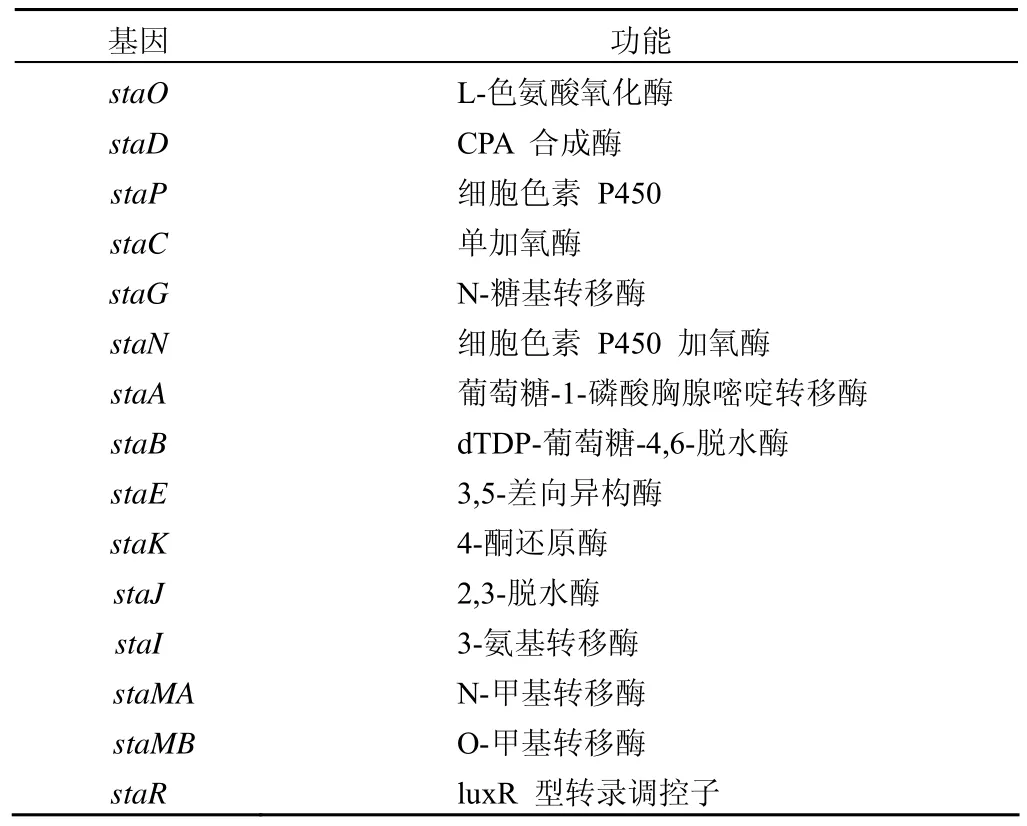
表 1 合成星形孢菌素的基因及其功能
星形孢菌素生物合成的一个关键步骤是在细胞色素P450 StaP 的催化下通过分子内 C-C 键的形成和 CPA 的氧化脱羧形成吲哚咔唑核心。
StaP 属于细胞色素 P450 酶家族(P450s)。P450s 广泛分布于植物、哺乳动物、昆虫和细菌中,催化类固醇激素的合成、药物代谢和许多其他重要的单加氧反应。近年来P450 StaP 引起了许多研究者的兴趣,不仅因为 STA 重要的抗肿瘤活性,更是由于 P450 StaP 在 STA 合成中特别的催化作用:芳基-芳基的连接和氧化脱羧,而不是常见的氧插入。StaP 反应体系包括铁氧还蛋白(ferredoxin)、黄素氧还蛋白 NADP+还原酶(flavodoxin NADP+-reductase)、烟酰胺腺嘌呤二核苷酸(磷酸)[NAD(P)H]和氧分子,在其催化下 CPA 形成三种不同氧化水平的吲哚咔唑化合物:K252c,arcyriaflavin A和 7-羟基-K252c。值得注意的是,在额外加入 StaC 时只形成一种产物:K252c(STA 的糖苷配基)[14]。Howard-Jones 和 Walsh[15]经过对芳基-芳基连接后非酶催化步骤的研究认为 StaC 对中间产物起拦截和重定向作用,从而只产生了 K252c。18O 同位素标记的实验表明 K252c 吡咯酮上的氧原子来自于氧气,两分子的氧气产生一分子的 K252c[14]。
通过对 StaP 晶体学的研究,得到了 CPA 结合和非结合时 StaP X 衍射晶体结构图[5]。从图中观察到,StaP 具有P450 的保守结构即十二个 α 螺旋(A ~ L)及其所特有的两个 β 折叠。StaP 不与底物结合时呈现一种开放的构型,暴露其活性位点,使 CPA 易于接近亚铁血红素周围的底物结合区。当 CPA 靠近时,底物结合区的部分氨基酸残基重新定位与 CPA 相互作用,同时疏水性残基 Trp-97、Phe-100和 Phe-403 也改变位置,优化底物和酶的相互作用。CPA接近 StaP 的过程中,两个羧基的氢键作用和吲哚环的T 型 π - π 键作用使底物进入 StaP 的底物结合腔。CPA 在StaP 的活性位点呈现一种扭曲的蝴蝶型,几乎垂直于亚铁血红素平面。
整体上看,CPA 是在 P450 StaP 的催化下失去两个质子形成环闭结构。根据这一现象,许多研究者对其反应机理提出了假说。Howard-Jones 和 Walsh[15]认为 StaP 催化C-C 形成起始于 CPA 两个吲哚环 C-H 上质子的转移,再通过一系列的非酶催化反应形成最终产物。通过分析CPA-StaP 复合物 X 衍射晶体结构图可知[5],CPA 中靠近亚铁血红素的吲哚基团周围的静电势与细胞色素 C 过氧化物酶(cytochrome c peroxidase,CcP)周围的类似,故推测,在 Cpd I(StaP)-CPA 中涉及了一个单电子还原的 Cpd II 和一个单电子氧化的 CPA(Cpd I 和 Cpd II 在CcP 中称为 CcP 复合物 I 和 CcP 复合物 II,分别是四价和三价铁离子与氧络合形成的复合物)。反应起始,两个吲哚环N-H 上各失去一个质子并伴随一个额外电子的转移,随后两个吲哚环 C-C 连接并发生 N-C-H 的互变异构形成最终产物。然而,由于 CPA 被 StaP 底物结合位点周围一系列氢键束缚着,使吲哚环上的 C-H 和 N-H 离 Cpd I 或 Cpd II 太远而不能直接发生反应,这就需要一个中介。Wang等[16]结合实验和 QM/MM 计算法提出了氢键三元组:Wat644-His250-Wat789(StaP 的底物结合处具有一个质子穿梭残基 His250,两个水分子 Wat644 和 Wat789)这三个组分可以把质子从固定的 CPA 转运到 Cpd I 的氧原子上。首先在三元组的帮助下发生“质子耦合的电子转移(PCET)”,即 CPA 失去一个电子到亚铁血红素的同时,邻近的一个吲哚环 N-H 失去一个质子。环闭(C-C 连接)的形成是在Wat644-Wat789 帮助下失去第二个也是最后一个电子,即发生“键形成耦合的电子转移(BFCET)”。随后的实验证明His250 不是反应发生所必需的,因为二元组Wat644-Wat789 在第一个电子转移时也可以执行催化作用(虽然效率只有 His250 存在时的 75%),从而得出二元组Wat644-Wat789 是此反应发生所必需的基本元素。芳基-芳基连接反应也存在于 P450 158A2 催化淡黄霉素(flaviolin)二聚体[17]形成及 P450 OxyC 催化万古霉素(vancomycin)的生物合成中[18]。
环闭合后是吡咯环的脱羧和氧化。空间位阻及静电相互作用利于脱羧反应的发生。通过芳基-芳基的连接,CPA的构象由扭曲的蝴蝶型转变为平面结构,进一步促进了CPA 的脱羧,避免羧基和平面吲哚咔唑核心的排斥。Howard-Jones 和 Walsh[15]实验证实,StaP 在 CPA 形成中所起的作用只是连接两个吲哚环(C-C),电子富集的吡咯环在有氧环境中不稳定,在溶液中可自发脱羧和氧化,并且限速步骤是随后的脱羧氧化而不是 C-C 连接。
2 星形孢菌素的外源表达
星形孢菌素最初发现于 Streptomyces Staurosporeus AM-2282。目前已经报道的可以产生 STA 的菌株包括Streptomyces sp. TP-A0274[5],Lentzea albida(formerly Streptomyces staurosporeus)[19],Streptomyces longisporo fl avus DSM10189[20],Lechevalieria aerocolonigenes subsp. Streptomyces Staurosporeus AM-2282[9],Stretomyce sp.4138[21]。
目前报道的可以产生 STA 的异源宿主有 Streptomyces albus[22],Streptomyces longisporo fl avus[22],Streptomyces lividans[6]。其中 Salas 等[22]在 S. albus 体内利用双质粒系统完整重建了 STA 的合成路径。第一个质粒(aglycone plasmid)含 STA 吲哚咔唑环合成基因(staO、staD、staP 和staC),STA 糖基转移酶 staG 基因和负责吲哚咔唑核心与糖残基第二个 C-N 连接形成的 staN 基因[23-24]。StaN 是P450s 的同系物,在此首次发现 P450 同系物参与 C-N 键形成[23]。另一个质粒(sugar plasmid)含氨基己糖合成基因(staE,staK,staJ,staI),有的还具有修饰糖残基的O-甲基转移酶和 N-甲基转移酶基因(staMA,staMB)。STA氨基糖合成的前两个步骤,由宿主 S. albus 提供的 StaA 和StaB 催化[22]。StaG 对于糖供体表现出一定的灵活性,能够转移多种脱氧己糖。在 S. albus 体内共表达这两种质粒产生了星形孢菌素和一些糖基化的衍生物。当替换 sugar plasmid 时,StaG 和 StaN 可以转移并连接这些糖基到吲哚咔唑环上,从而产生多种新型糖基化衍生物[25],包括4 种单键连接的衍生物,左旋鼠李糖(L-rhamnose)、左旋橄榄糖(L-olivose)、左旋洋地黄毒素糖(L-digitoxose)、右旋橄榄糖(D-olivose)或 3 种双键连接的衍生物(3 种 L 型糖形成的双键化合物)。在第二个 C-N 键连接时,糖残基需要从4C1-构象转变为1C4-构象,而 D 型糖基可能由于热力学和空间位阻效应,阻碍了第二个 C-N 键的形成。故双C-N 连接只在 L 型糖中发生[22]。
星形孢菌素在异源宿主体内表达产量没有在原宿主体内表达量高。STA 表达量在异源宿主 Streptomyces lividans 体内是 2.6 mg/L(培养 9 d),而在原宿主体内是10.5 mg/L[9]。
Cai 等[26]推测,STA 在微生物体内处于甲酰化状态。由于 STA 具有细胞毒性,当其在菌体内产生时,可能被甲酰化从而保护菌体不受自身代谢产物的毒害,当分泌到体外后去甲酰基得到 STA。
3 星形孢菌素的生物功能及作用机制
STA 具有多种生物学功能,包括抗真菌,降血压[27],促血小板聚集等,其中最重要的是作为激酶抑制剂的抗肿瘤活性[3]。STA 与这些激酶的 ATP 结合域相互作用,这就解释了为什么 STA 及其类似物(糖基与吲哚咔唑核心双连接)的抑制活性缺乏选择性。平面六环框架和碳水化合物部分在识别细胞内目标物时均具有重要作用。在 STA-蛋白激酶结构中,糖苷配基部分恰好进入蛋白激酶的 ATP 结合域,大致叠合在 ATP 的嘌呤位置上,吲哚氮原子和糖基氢氧根形成关键的氢键[28]。
进一步的生化研究表明 STA 与 PKC 的催化区域结合,而 PKC 的催化区域与其他激酶的催化区域具有很大的相似性,这导致 STA 对激酶的选择性极差,在抑制 PKC的同时也能抑制其他蛋白激酶。STA 对 PKC 异构酶的选择性则几乎没有。
鉴于星形孢菌素的高活性,低选择性,人们很自然以它作为先导化合物对其进行结构改造希望能找到选择性的PKC 拮抗剂。通过对 STA 的改造,人们发现了第一个在体外能选择性地拮抗 Ca2+依赖性与 Ca2+不依赖性的PKC 异构酶的化合物 GOE6976。随后几年又改造得到了多种具有潜在抗肿瘤活性的 STA 衍生物,如 NB-506 和BMY-27557。
4 展望
星形孢菌素是非常有效的蛋白激酶抑制剂,但缺乏选择性,如果作为药物,离人们预期还有很大距离。天然产物是药物先导物的可靠来源,故研究者常以对 PKC 具有拮抗能力的天然化合物为先导物进行结构改造来获得选择性的PKC 拮抗剂。例如将 STA 的吲哚咔唑结构改为 2,3-双吲哚马来酰亚胺结构,使得化合物的选择性提高。而从一些吲哚咔唑化合物产生菌中分离得到生物合成基因簇并应用组合生物合成技术(体内合成途径)在异源宿主体内表达,得到的 STA 衍生物比生物转化带有细胞毒性的 STA 更有效,在增加化合物结构多样性方面具有广阔的应用前景,为产生高效的有选择性的蛋白激酶抑制剂提供潜在可能。
对比于其他复杂天然产物的合成路径,吲哚咔唑的合成途径相对简单,并且一些催化吲哚咔唑化合物合成的酶类具有一定程度的底物灵活性,利于产生多种新的衍生物。因此,在保持 STA 吲哚咔唑核心不变的情况下可以对糖基进行改造,例如当糖基连接到糖苷配基上以后可以进一步进行后糖基化修饰,例如甲基化或酰化(STA 就是连接后甲基化产生)。另外,一种作为对体外糖类随机化(IVG)补充的糖类随机化新方法——新型糖类随机化(neoglycorandomization)在制药方面有巨大的潜力。已经证明,此法可以提高强心苷的抗肿瘤活性[29-30]。
不论是用化学方法还是用生物合成方法对 STA 的吲哚咔唑核心或是糖残基改造,都需要将两部分连接,因此,糖基转移酶是不可或缺的。只有对吲哚咔唑类糖基转移酶结构认识不断深入才能提高 STA 衍生物的合成效率。
星形孢菌素市场价格昂贵,目前只用于医学领域进行理论研究。可以通过选育来提高产生菌的发酵效价,构造更稳定的菌株,从而真正扩大生产用于制药工业。
[1] Funato N, Takayanagi H, Konda Y, et al. Absolute configuration of staurosporine by X-ray analysis. Tetrahedron Lett, 1994, 35(8):1251-1254.
[2] Tamaoki T, Nomoto H, Takahashi I, et al. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem Biophys Res Commun, 1986, 135(2):397-402.
[3] Link JT, Raghavan S, Danishefsky SJ. First total synthesis of staurosporine and ent-staurosporine. J Am Chem Soc, 1995, 117(1):552-553.
[4] Salas JA, Méndez C. Indolocarbazole antitumour compounds by combinatorial biosynthesis. Curr Opin Chem Biol, 2009, 13(2):152-160.
[5] Makino M, Onaka H, Nagano S, et al. Crystal structures and catalytic mechanism of cytochrome P450 StaP that produces the indolocarbazole skeleton. Proc Natl Acad Sci U S A, 2007, 104(28):11591-11596.
[6] Onaka H, Taniguchi S, Igarashi Y, et al. Cloning of the staurosporine biosynthetic gene cluster from streptomyces sp.TP-A0274 and its heterologous expression in Streptomyces lividans. J Antibiot (Tokyo),2002, 55(12):1063-1071.
[7] Sánchez C, Salas AP, Braña AF, et al. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb), 2009, (27):4118-4120.
[8] Asamizu S, Kato Y, Igarashi Y, et al. Direct formation of chromopyrrolic acid from indole-3-pyruvic acid by StaD, a novel hemoprotein in indolocarbazole biosynthesis. Tetrahedron Lett, 2006,47(4):473-475.
[9] Onaka H. Biosynthesis of heterocyclic antibiotics in actinomycetes and an approach to synthesize the natural compounds.Actinomycetologica, 2006, 20(2):62-71.
[10] Onaka H, Taniguchi S, Igarashi Y, et al. Characterization of the biosynthetic gene cluster of rebeccamycin from Lechevalieria aerocolonigenes ATCC 39243. Biosci Biotechnol Biochem, 2003,67(1):127-138.
[11] Sánchez C, Zhu L, Braña AF, et al. Combinatorial biosynthesis of antitumor indolocarbazole compounds. Proc Natl Acad Sci U S A,2005, 102(2):461-466.
[12] Wang Y, Hirao H, Chen H, et al. Electron transfer activation of chromopyrrolic acid by cytochrome p450 en route to the formation of an antitumor indolocarbazole derivative: theory supports experiment.J Am Chem Soc, 2008, 130(23):7170-7171.
[13] Onaka H. Biosynthesis of indolocarbazole and goadsporin, two different heterocyclic antibiotics produced by actinomycetes. Biosci Biotechnol Biochem, 2009, 73(10):2149-2155.
[14] Howard-Jones AR, Walsh CT. Staurosporine and rebeccamycin aglycones are assembled by the oxidative action of StaP, StaC, and RebC on chromopyrrolic acid. J Am Chem Soc, 2006, 128(37):12289-12298.
[15] Howard-Jones AR, Walsh CT. Nonenzymatic oxidative steps accompanying action of the cytochrome P450 enzymes StaP and RebP in the biosynthesis of staurosporine and rebeccamycin. J Am Chem Soc, 2007, 129(36):11016-11017.
[16] Wang Y, Chen H, Makino M, et al. Theoretical and experimental studies of the conversion of chromopyrrolic acid to an antitumor derivative by cytochrome P450 StaP: the catalytic role of water molecules. J Am Chem Soc, 2009, 131(19):6748-6762.
[17] Zhao B, Guengerich FP, Bellamine A, et al. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J Biol Chem,2005, 280(12):11599-11607.
[18] Pylypenko O, Vitali F, Zerbe K, et al. Crystal structure of OxyC, a cytochrome P450 implicated in an oxidative C-C coupling reaction during vancomycin biosynthesis. J Biol Chem, 2003, 278(47):46727-46733.
[19] Yang SW, Cordell GA. Origin of nitrogen in the indolocarbazole unit of staurosporine. J Nat Prod, 1997, 60(8):788-790.
[20] Schupp T, Engel N, Bietenhader J, et al. (1997) Staurosporin biosynthesis gene clusters. World Intel-lectual Property Organization Patent WO9708323.
[21] Pu XM, Lin BR, Hu MY, et al. Screening of staurosporine-producing mutant strain. J Nucl Agric Sci, 2008, 22 (3):276-279. (in Chinese)蒲小明, 林壁润, 胡美英, 等. 星形孢菌素产生菌的选育研究. 核农学报, 2008, 22(3):276-279.
[22] Salas AP, Zhu L, Sánchez C, et al. Deciphering the late steps in the biosynthesis of the anti-tumour indolocarbazole staurosporine: sugar donor substrate fl exibility of the StaG glycosyltransferase. Mol Microbiol, 2005, 58(1):17-27.
[23] Onaka H, Asamizu S, Igarashi Y, et al. Cytochrome P450 homolog is responsible for C-N bond formation between aglycone and deoxysugar in the staurosporine biosynthesis of Streptomyces sp.TP-A0274. Biosci Biotechnol Biochem, 2005, 69(9):1753-1759.
[24] Sánchez C, Méndez C, Salas JA. Engineering biosynthetic pathways to generate antitumor indolocarbazole derivatives. J Ind Microbiol Biotechnol, 2006, 33(7):560-568.
[25] Rodríguez L, Aguirrezabalaga I, Allende N, et al. Engineering deoxysugar biosynthetic pathways from antibiotic-producing microorganisms. A tool to produce novel glycosylated bioactive compounds. Chem Biol, 2002, 9(6):721-729.
[26] Cai Y, Fredenhagen A, Hug P, et al. Further minor metabolites of staurosporine produced by a Streptomyces longisporoflavus strain. J.Antibiot (Tokyo), 1996, 49(6):519-526.
[27] Furusaki A, Hashiba N, Matsumoto T, et al. X-Ray crystal structure of staurosporine :a new alkaloid from a Streptomyces strain. J Chem Soc Chem Commun, 1978, 18:800-801.
[28] Prade L, Engh RA, Girod A, et al. Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential. Structure, 1997, 5(12):1627-1637.
[29] Langenhan JM, Peters NR, Guzei IA, et al. Enhancing the anticancer properties of cardiac glycosides by neoglycorandomization. Proc Natl Acad Sci U S A, 2005, 102(35):12305-12310.
[30] Salas JA, Méndez C. Engineering the glycosylation of natural products in actinomycetes. Trends Microbiol, 2007, 15(5):219-232.
猜你喜欢
食品与生物技术学报(2022年1期)2023-01-11
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
科学(2020年2期)2020-08-24
中国免疫学杂志(2019年17期)2019-09-20
分析化学(2017年5期)2017-06-21
中国塑料(2015年10期)2015-10-14
医学研究杂志(2015年3期)2015-06-10
中国洗涤用品工业(2014年12期)2014-01-30
