
*10-羟基苯并喹啉锂配合物电子结构和光谱性质的理论研究
2011-01-11 08:21:28李杰贺婧媛程瑛徐雷戴键鑫邢立文康延赏施和平
山西大学学报(自然科学版) 2011年1期
李杰,贺婧媛,程瑛,徐雷,戴键鑫,邢立文,康延赏,施和平
(1.山西大学化学化工学院,山西太原 030006;2.忻州师范学院五寨分院,山西五寨 036200)
*10-羟基苯并喹啉锂配合物电子结构和光谱性质的理论研究
李杰1,2,贺婧媛1,程瑛1,徐雷1,戴键鑫1,邢立文1,康延赏1,施和平1*
(1.山西大学化学化工学院,山西太原 030006;2.忻州师范学院五寨分院,山西五寨 036200)
采用B3LYP和ab initio CIS方法在6-31G(d,p)水平上对10-羟基苯并喹啉锂配合物(LiBQ配合物)的基态和激发态的几何构型进行全优化.在基态和激发态的优化构型的基础上采用含时密度泛函理论(TD-DFT)的PCM模型计算配合物的吸收和发射光谱.计算得到的最大吸收和最大发射都是由于配体内电荷转移(ILCT)所致.计算结果表明配合物(LiBQ)是一种良好的发射蓝光的金属有机电致发光材料.
10-羟基苯并喹啉锂配合物;TD-DFT;吸收光谱;发射光谱
自1987年 Tang等[1]发现三(8-羟基喹啉)铝(III)(A lq3)是具有高量子产率的金属有机电子传输材料以来,基于金属有机配合物的电致发光技术的研究已经成为当前研究的热点.研究人员致力于合成出性能更优良、更稳定、且具有特定发射光谱的新的金属有机配合物[2].从某种意义上讲,有机配体的性能直接影响金属有机配合物的电致发光的性能,而有机配体的性能又主要取决于有机分子的结构.
近年来,密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)已经被人们广泛接受.用来描述金属有机配合物基态、激发态的性质和金属有机配合物的电致发光性能.目前,国内外的研究组在配合物光谱性质的量子化学研究方面,取得了相当多的研究结果[3-14].
在本文中,我们报道10-羟基苯并喹啉锂配合物(LiBQ配合物)的电子结构和光谱性质的理论研究,以便更深入地了解这种配合物的结构和电致发光性能.
1 计算方法
首先,用B3L YP/6-31G(d,p)方法对LiBQ配合物的基态构型进行优化[15-17],在得到优化构型的基础上分析振动频率和前线轨道特征.然后,用B3L YP/6-31G(d,p)的方法对离子的几何结构进行优化,在此基础上计算LiBQ配合物的电离能和电子亲和能.最后,在6-31G(d,p)水平上采用单重激发态(CIS)方法对LiBQ配合物的激发态几何构型进行优化.在优化构型的基础上,采用含时密度泛函理论(TD-DFT)计算LiBQ配合物在CH2Cl2溶液的吸收光谱和发射光谱[18].所有计算均使用 Gaussian 03程序包[19],在中科院超级计算中心的深腾7 000超级计算机上完成.
2 结果和讨论
2.1 LiBQ配合物的基态的结构
用DFT/B3LYP/6-31G(d,p)方法对LiBQ配合物进行全优化.LiBQ配合物的优化的结构见图1(P102).优化后得到的几何参数见表1(P102).由图1和表1可以看出,优化后的基态结构是平面结构.

图1 基态时LiBQ配合物的优化结构图Fig.1 Optimized geometry of LiBQ comp lex in the ground state
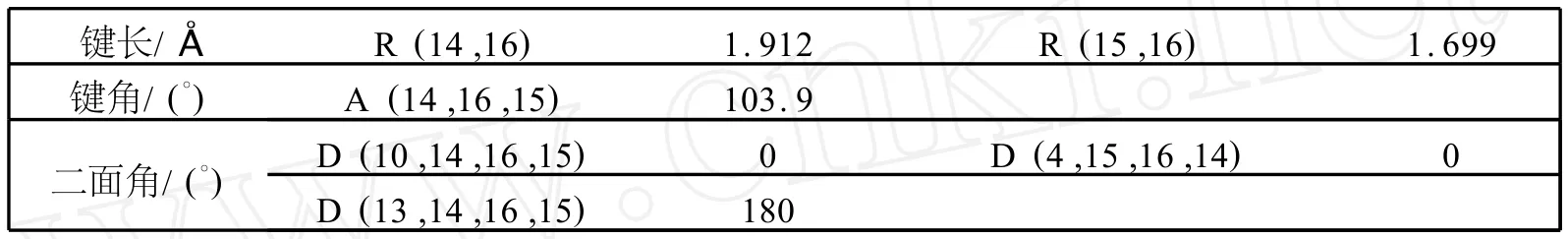
表1 在B3L YP/6-31G(d,p)下,LiBQ的基态的部分优化的键长,键角和二面角Table 1 Selected optimized bond lengths,bond angles and dihedral angles of the L iBQat B3LYP/6-31G(d,p)level in their ground state
2.2 基态下LiBQ配合物的前线分子轨道性质
前线分子轨道是衡量化合物的激发特性和电子或空穴传输能力的一个合理的定性指标[20].LiBQ配合物的HOMO和LUMO示意图见图2.从图2中可以看出LiBQ配合物的HOMO的电荷密度主要在配体苯酚环上.这是成键轨道.LiBQ配合物的LUMO的电荷密度主要在吡啶环上.这是反键轨道.电子从基态跃迁到激发态主要是由含氧的苯酚环流向含氮的吡啶环,属于π→π*跃迁.金属Li离子起到支持和约束配体的作用.它对 HOMO和LUMO没有贡献,不直接参与发光.
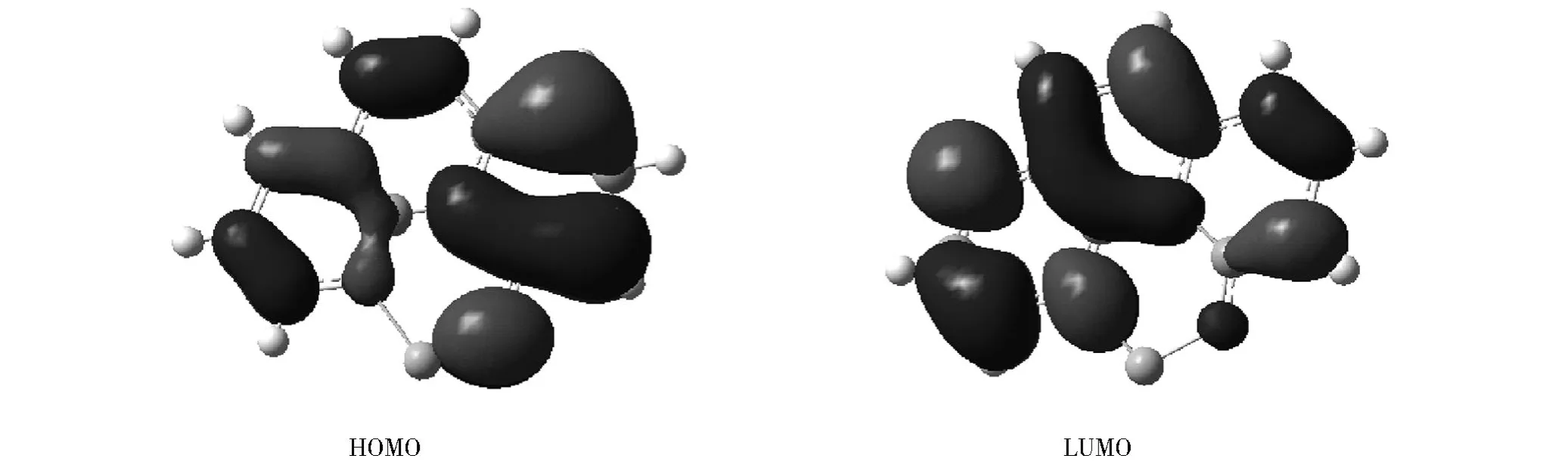
图2 基态下LiBQ配合物的 HOMO和LUMO图Fig.2 Isodensity surface p lots of HOMO and LUMO of LiBQ comp lex in the ground state
2.3 基态下LiBQ配合物的电离能和电子亲和能
电离能(IP)包括垂直电离能(IPv)和绝热电离能(IPa).垂直电离能是在中性分子的优化构型基础上,阳离子与中性分子的能量的差.而绝热电离能是优化的阳离子与优化的中性分子的能量差.电子亲和能(EA)包括垂直电子亲和能(EAv)和绝热电子亲和能.垂直电子亲和能是在中性分子的优化构型基础上,阴离子与中性分子的能量的差,而绝热电子亲和能是优化的阳离子与优化的中性分子的能量差.LiBQ配合物的IP和EA列于表2(P103).从表2可知在基态下LiBQ配合物的电离能是6.280 eV和6.137 eV,电子亲和能是0.244 eV和0.079 eV.LiBQ配合物有较大的电离能可以阻挡空穴,同时又有较小的电子亲和能有利于电子传输.

表2 基态下LiBQ配合物的电离能和电子亲和能Table 2 IP and EA of the LiBQ complex in the ground state(eV)
2.4 基态下LiBQ配合物的电子吸收光谱
在用B3L YP/6-31G(d,p)方法得到的基态(S0)优化构型基础上,采用含时密度泛函理论(TD-DFT)计算了LiBQ配合物在CH2Cl2溶液中的电子吸收光谱.计算数据列于表3.以计算所得的垂直激发能和相应的振子强度为基础,借助于SW IZARD软件模拟吸收光谱.LiBQ配合物模拟的吸收光谱见图3.从图3和表3中可以看出LiBQ配合物的最大吸收波长为413 nm.在CH2Cl2溶剂中,计算的最大波长是由 HOMO到LUMO的电子转移引起的.他们主要是由于具有配体内电荷转移(IL TC)特征的π{(苯酚)→π*(吡啶)}转移引起的.

图3 模拟的LiBQ配合物在CH2 Cl2溶液中的吸收光谱Fig.3 Simulated absorption spectrum of LiBQ complex in CH2 Cl2 sovent

图4 LiBQ配合物的第一单重激发态的HOMO和LUMO图Fig.4 Contour p lots of HOMO and LUMO of LiBQ comp lex in the first excited state

表3 计算的LiBQ配合物的电子吸收光谱数据Table 3 Computed electronic absorption spectrum data of LiBQ complex in the ground state
2.5 LiBQ配合物的第一激发态的优化结构
用单重激发态(CIS)方法在6-31G(d,p)基组上计算得到了LiBQ配合物的第一单重激发态(S1)的优化构型.优化后的主要几何结构参数列于表4.第一单重激发态的 HOMO和LUMO见图4.LiBQ配合物优化后的第一单重激发态结构类似于基态结构.
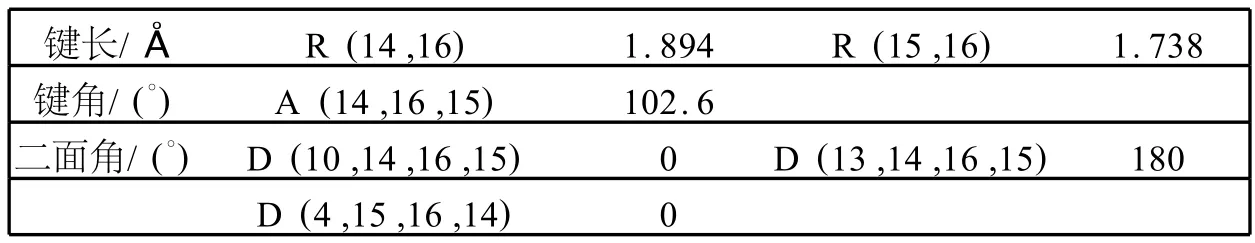
表4 第一激发态下LiBQ配合物部分优化的键长,键角和二面角Table 4 Selected optimized bond lengths,bond angles and dihedral angles of LiBQ complex in the first excited state
2.6 LiBQ配合物的发射光谱
首先采用 HP/6-31G(d,p)方法得到LiBQ配合物的第一激发态(S1)的优化结构,随后在所得到的优化结构的基础上,采用含时密度泛函理论(TD-DFT)方法计算其在CH2Cl2溶液中的电子发射光谱.计算数据列于表5.LiBQ配合物模拟的发射光谱见图5.从表5中可以看出,LiBQ配合物的发射峰来源于具有激发态配体内电荷转移(IL TC)特征的π{(苯酚)→π*(吡啶)}转移.LiBQ配合物在CH2Cl2溶剂中最大发射波长为430 nm.计算结果表明,LiBQ配合物是一种具有发射蓝光的有机电致发光材料.
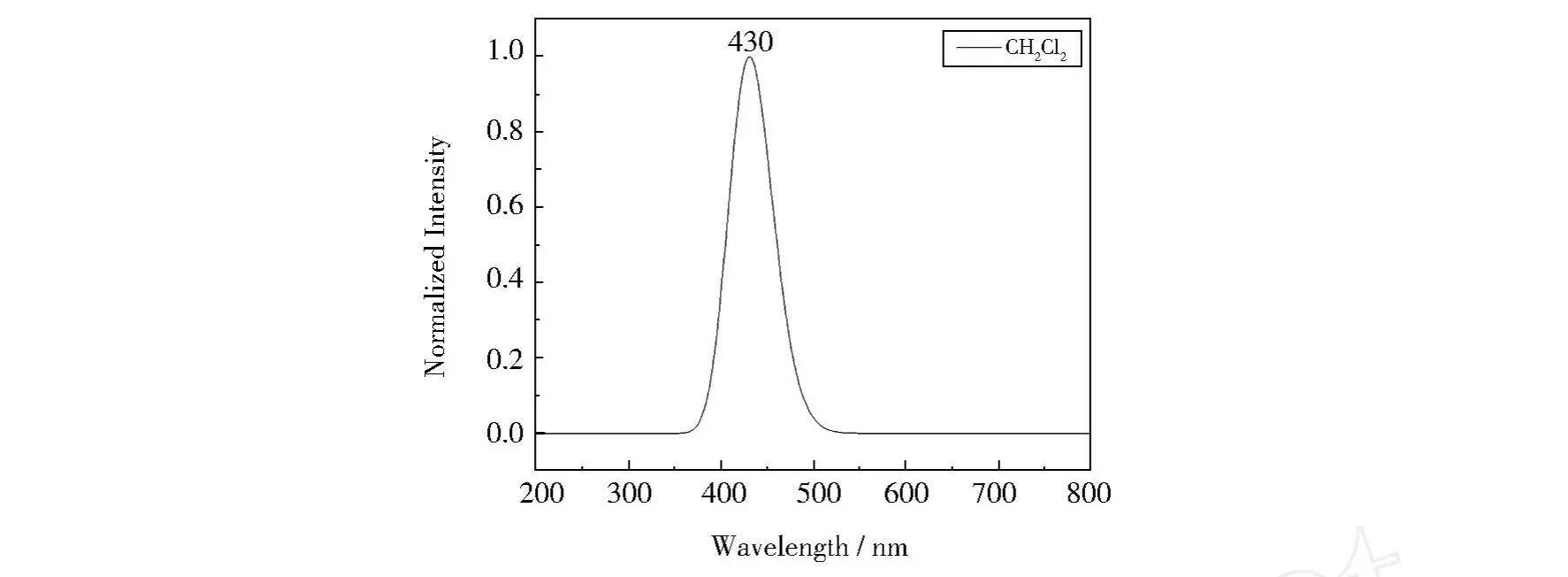
图5 第一激发态下模拟的LiBQ配合物的发射光谱Fig.5 Simulated emission spectrum of LiBQ complex in the ground state
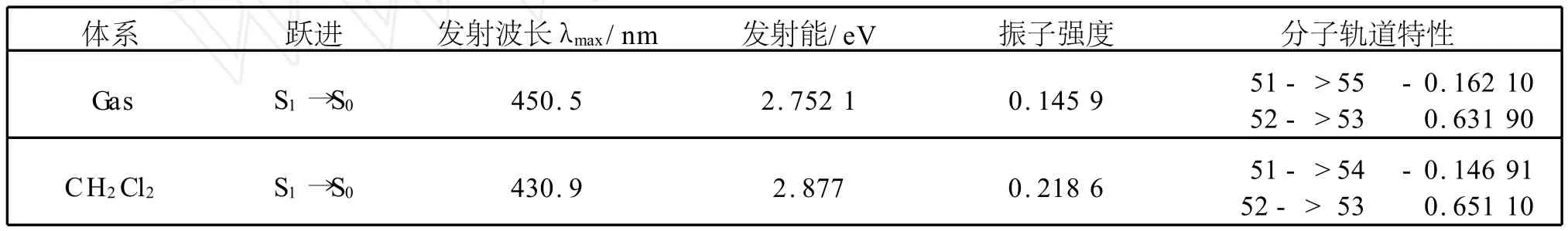
表5 第一激发态下计算的LiBQ配合物计算的发射光谱数据Table 5 Computed emission spectrum data of LiBQ complex in the first excited state
3 结论
本论文基于密度泛函理论(DFT)和单重激发态(CIS)方法在6-31G(d,p)水平上,对LiBQ配合物进行了理论研究.采用TD-DFT方法计算了配合物在CH2Cl2溶液中的吸收和发射光谱.计算结果表明配合物的最大吸收主要归结于配体内电荷转移(ILCT),最大发射也主要来自于配体内电荷转移(ILCT).研究结果表明该配合物是良好的电致发光材料,该研究为金属有机电致发光材料的分子结构设计提供了一定的理论依据.
[1] Tang CW,VanSlyke S A.Organic Electroluminescent Diodes[J].ApplPhys Lett,1987,51:913-915.
[2] Ghedini M,LaDeda M,Aiello I,et al.Synthesis and Photophysical Characterisation of Soluble Photoluminescent Metal Complexes with Substituted 8-hydroxyquinolines[J].Synthetic M et,2003,138:189-192.
[3] Jamo rski C,Casida M E,Salahub D R.Dynamic Polarizabilities and Excitation Spectra from a Molecular Imp lementation of Time-dependent Density-functional Response Theory:N2 as a Case Study[J].J Chem Phys,1996,104:5134-5147.
[4] Stratmann R E,Scuseria G E,Frisch M J.An Efficient Implementation of Time-dependent Density-functional Theo ry fo r the Calculation of Excitation Energies of Large Molecules[J].J Chem Phys,1998,109:8218-8224.
[5] Rosa A,Baerends E J,VanGisbergen SJ A A,et al.Electronic Spectra of M(CO)6(M=Cr,Mo,W)Revisited by a Relativistic TDDFT App roach[J].J Am Chem Soc,1999,121:10356-10365.
[6] Adamo C,Barone V.Inexpensive and Accurate Predictions of Optical Axcitations in Transition-metal Complexes:the TDDFT/PBEO route[J].Theor Chem Acc,2000,105:169-172.
[7] Farrell IR,VanSlageren J,Zalis S,et al.Time-resolved Emission Spectra and TD-DFT Excited-state Calculations of[W(CO)4(1,10-phenanthroline)]and[W(CO)4(3,4,7,8-tetramethyl-1,10-phenanthroline)][J].Inorg Chim Acta,2001,315:44-52.
[8] Hay PJ.Theoretical Studies of the Ground and Excited Electronic States in Cyclometalated Phenylpyridine Ir(III)Complexes Using Density Functional Theory[J].J Phys Chem,2002,A 106:1634-1641.
[9] Halls M D,Schelgel H B.Molecular O rbital Study of the First Excited State of the OLED Material Tris(8-hydroxyquinoline)aluminum(III)[J].Chem M ater,2001,13:2632-2640.
[10] 李明霞,周欣,张红星,等.[M(N)X2](M=Ru,O s;X=S2C6H4,mnt)的电子结构和光谱性质的理论研究[J].高等学校化学学报,2009,30(11):2284-2287.
[11] 吴玉辉,周欣,王嵩,等.IrR(CO)(PH3)2(mnt)配合物的基态和激发态结构及光谱性质的理论研究[J].化学学报,2009,67(1):33-38.
[12] 吴玉辉,周欣,张红星.系列二亚胺Os(II)配合物[Os(L)2(CN)2(phen)](L=PH3,DMSO;phen=1,10-邻二氮杂菲)及[Os(PH3)2(phen)Br2]电子结构和光谱性质的理论研究[J].化学学报,2009,67(3):197-202.
[13] 廖奕,苏忠民,陈亚光,等.8-羟基喹啉铍及其衍生物电子光谱性质的含时密度泛函理论研究[J].高等学校化学学报,2003,24(3):477-480.
[14] 颜力楷,苏忠民,仇永清,等.水杨醛缩乙二胺双席夫碱及其Ni(Ⅱ)配合物的电子结构和非线性光学性质的INDO/C I研究[J].高等学校化学学报,2006,27(4):711-715.
[15] Becke A D.Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior[J].Phys Rev A,1988,38:3098-3100.
[16] Becke A D.Density-functional thermochemistry.III.The Role of Exact Exchange[J].J Chem Phys,1993,98:5648-5652.
[17] Lee C,Yang W,Parr R G.Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density[J].Phys Rev,1988,B37:785-789.
[18] Casida M E,Jamorski C,Casida K C,et al.Molecular Excitation Energies to High-lying Bound States from Timedependent Density-functional Response Theory:Characterization and Correction of the Time-dependent Local Density Approximation Ionization Threshold[J].J Chem Phys,1998,108:4439-4449.
[19] Frisch M J,Trucks GW,Schlegel H B,et al.Gaussian 03[CP].Gaussian Inc,Pittsburgh,PA,2003.
[20] Belletete M,Mo rin J F,Leclerc M,et al.A Theo retical,Spectroscopic,and Photophysical Study of 2,7-Carbazolenevinylene-Based Conjugated Derivatives[J].J Phys Chem,2005,A 109:6953-6959.
Theoretical Study on Electronic Structure and Spectra
Properties of 10-hydroxybenzo[h]quinoline L ithium Complex
L IJie1,2,HE Jing-yuan1,CHENG Ying1,XU Lei1,DA IJian-xin1,XING Li-wen1,KANG Yan-shang1,SH IHe-ping1
(1.School of Chemistry and Chem ical Engineering,Shanxi University,Taiyuan030006,China;2.Wuzhai B ranch of Xinzhou Teachers College,W uzhai036200,China)
The geometric structures of 10-hydroxybenzo[h]quinoline lithium(LiBQ)comp lex in the groundstate(S0)and the low est singlet excited-state(S1)were optimized by B3L YP and ab initio CIS methods at 6-31G(d,p)level respectively.The absorption and emission spectra of the comp lex were investigated by the time dependent density functional theory(TD-DFT)level with PCM model on the basis of the optimized ground and excited states geometry respectively.The calculated lowest-lying absorption of the comp lex was attributed to intraligand charge transfer(ILCT)transitions mainly.The calculated emission of the comp lex can be described as originated from an excited state with intralig and charge transfer(ILCT)character mainly.The result displayed that the comp lex w as a potential electroluminescent material with blue light emission.
10-hydroxybenzo[h]quinoline lithium;TD-DFT;absorption spectrum;emission spectrum
O627
A
0253-2395(2011)01-0101-05*
2010-06-22;
2010-09-14
山西省科技项目(20100321085)
李杰(1972-),男,山西五寨人,硕士研究生,山西省忻州师范学院五寨分院教师,主要从事应用化学.E-mail:hepingshi@sxu.edu.cn
猜你喜欢
纺织科学研究(2023年12期)2023-12-19 12:36:08
——以物质结构与性质模块“元素周期律”教学为例
化学教与学(2023年3期)2023-02-09 08:33:22
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
读与写·上旬刊(2018年10期)2018-11-27 20:03:08
中央民族大学学报(自然科学版)(2016年2期)2016-06-27 01:29:06
原子与分子物理学报(2015年3期)2015-11-24 12:49:36
化学教与学(2014年6期)2014-07-03 10:04:09
原子与分子物理学报(2014年1期)2014-03-20 08:16:14
计算物理(2014年2期)2014-03-11 17:01:44
