
FFC法制备金属钛研究进展
2010-12-25 07:51唐仁衡肖方明
材料研究与应用 2010年4期
李 伟,王 英,唐仁衡,肖方明
(广东省工业技术研究院(广州有色金属研究院)稀有金属研究所,广东 广州 510650)
FFC法制备金属钛研究进展
李 伟,王 英,唐仁衡,肖方明
(广东省工业技术研究院(广州有色金属研究院)稀有金属研究所,广东 广州 510650)
对熔盐中直接电还原固态二氧化钛制备金属钛(FFC法)的基本原理进行了介绍,综述了近期相关研究动态和工业化进程,分析其实现工业化所需解决的问题,指出该工艺是目前金属钛及其合金制备工艺中最引入注目的一种方法,并且其工业化的实现指日可待.
FFC-剑桥工艺;二氧化钛;熔盐;钛
1 前 言
近年来,作为二氧化碳排放大户的汽车和其他运输行业,为了降低汽车重量以节能,并满足更为严格的排放标准,开始逐步用钛及钛合金材料替换传统以钢为主的汽车部件和结构件.但是,钛及钛合金的应用必须解决成本问题.目前海绵钛的生产采用Kroll法[1].该方法自从1948年开发当初就因其成本高、还原效率低而受到批评.半个世纪过去了,该工艺并没有根本的改变,仍然是间歇式生产,未能实现连续化,生产成本很高,每千克钛所需能耗约为45~55 k Wh.同时,在该工艺中必须经过TiO2到TiCl4的转化,其反应式如下:TiO2(金红石)+2C+2Cl2(g)=TiCl4(g)+2CO(g).而产生的CO最终继续氧化为CO2.因此,该方法在高能耗的同时,还存在严重的CO2排放问题(2.04 kg-CO2/kg-Ti).
早在19世纪初,熔盐电解法已成功用于Mg、Al及Na、Li等碱金属或碱土金属的生产,许多稀有金属也可以用熔盐电解法制备.与其他方法相比,采用熔盐电解法制取金属有以下优点:1、较宽的电化学窗口,可允许电正性较强的金属(如钛)沉积;2、较高的温度使电化学反应动力学加快,单位时间内的产量也将提高;3、熔盐导电性好,能耗低;4、反应物在熔盐中溶解度高,浓度大,阴极电流密度高.因此,Kroll法的发明者Kroll就曾在20世纪50年代预言,大约需要15年的时间,熔盐电解法就有可能取代Kroll法生产金属钛,从而降低钛的价格,就像电解铝降低铝的价格一样.但遗憾的是直到今天这个预言尚未成为现实.不少学者开展了电解方法制钛的研究,但是都没有成功,电解法始终处于实验室阶段,未能实现工业化生产.
2000年9月,Chen等报道了在CaCl2熔盐中直接电解固态TiO2为金属钛的研究结果[2],引起各国学术界和工业界的普遍关注.工业上称其为FFC剑桥工艺(FFC Cambridge Process),并认为该方法可以大幅度降低金属钛及其合金的生产成本,是一种过程简单、成本较低、环境友好的全新材料生产方法,是目前各种金属合金提取工艺中最引人注目的一种方法.
2 FFC法工艺简介
FFC剑桥工艺具有过程简单、污染少和可连续化生产等优点,是一种新型绿色低碳短流程冶金工艺,其技术核心是将固态氧化物制成阴极并在低于金属熔点的温度和熔盐分解的电压下电解.即在CaCl2熔盐中以固态氧化物为阴极,以石墨(或惰性材料)为阳极,在低于金属熔点温度和熔盐分解电压下电解,金属氧化物被还原成金属或者合金,而氧以离子化形式进入熔盐迁移到阳极放电,生成气体(CO,CO2或O2).具体工艺流程如下:首先将 TiO2粉末用压铸或浇注的方法成型,烧结后用耐高温的金属集流体连接或包裹作为阴极,以石墨作为阳极,在850~950℃CaCl2熔盐体系中电解.电解过程中,固态TiO2阴极中氧离子化后迁出氧化物,通过CaCl2熔盐到达阳极放电,产生O2(惰性阳极)或者CO及CO2气体(石墨阳极),阴极则形成金属钛[3].图1为FFC法的工艺流程示意图,其基本反应如式(1)~(4).

图1 FFC法的工艺流程示意图
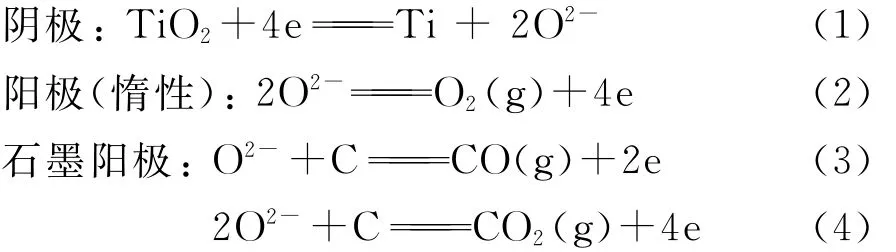
3 FFC法制备金属钛研究进展
据公开文献报道[4-5],该方法可以在3.0~3.2 V的电解电压下,将TiO2较快地还原为金属钛,但需要较长电解时间以降低金属钛中的固溶氧含量.由于熔盐的本征电子导电等因素的影响[6-7],长时间电解决定了目前大多数实验室在研究FFC方法过程中得到的低电流效率(氧含量(质量分数)0.2%vs.电流效率15%).尽管如此,FFC方法的能耗仍然只有33 k Wh/kg-Ti左右,比Kroll法低很多.显然,进一步提高电解效率可以加速实现FFC剑桥工艺的工业应用,而如何在较短的时间内电解获得低氧含量产品是关键.国内外众多研究者对FFC法的实验技术、TiO2的还原机理.动力学发展过程、电解工艺条件优化都进行了深入研究,以期推动该工艺的工业化进程.
3.1 实验技术
在熔盐体系中研究氧化物的还原机理,对实验技术及手段提出了高的要求.在氯化物熔盐电解研究过程中,大都采用恒槽压电解的方式,偶尔也使用假参比电极恒电位电解的方式.在高温氯化物熔盐中,恒电位电解比恒槽压电解更能实现反应的精确进行.比如采用FFC法制备高纯硅,由于极易产生硅钙合金,为了获得高纯硅必须采用参比电极严格控制电位进行[8].恶劣的高温环境以及氯化物熔盐的强腐蚀性,都对参比电极材料提出了苛刻的要求,因而制作通用的参比电极较为困难.高沛等[9-10]研究实现石英套管的局部固态离子导通,并由此设计了两种全密封的Ag/AgCl参比电极.这两类参比电极均具有制作简单,稳定性好(48 h电位漂移不超过8 m V),重现性佳,使用寿命长(连续使用72 h以上),不污染研究体系,可在多种氯化物熔盐体系中反复使用等特点.
为了研究熔盐中氧化物粉末的电化学行为,以及结合恒电位电解分析氧化物的具体还原机理,研究者把粉末微电极的优势运用于高温熔盐体系中[11].由于室温常用的粉末微电极难以在高温氯化物熔盐体系中使用,蒋凯等[12]设计了可适用于高温熔盐的钼金属微坑电极,具有使用方便、易于定性等特点,将该电极用于研究TiO2的还原取得了较理想的结果.邱国红等[13]设计的“金属通腔电极”,不仅可大大提高定量能力,而且便于在电化学测试前后作电子显微镜观察和成分分析,从而有利于机理研究.
3.2 TiO2 还原机理
关于FFC法氧化物阴极的还原机理存在氧离子 化[2]和 钙 热 还 原[14-15]两 种 争 议.Ono 和 Suzuki[14-15]等在 CaCl2熔盐中通过现场制备金属 Ca还原固态氧化物的方法制备得到了金属Ti、Nb、Ta等.认为氧化物阴极发生的反应如下:

2002年Chen等[16]采用循环伏安法研究了固态TiO2在Ca析出前的电化学行为,指出在Ca析出前TiO2就发生还原过程.K.Dring等[17]通过循环伏安曲线及XRD分析电解产物成分进一步确定了TiO2的还原机理:TiO2→Ti3O5→Ti2O3→TiO→Ti,其中TiO2与在附近CaCl2熔盐中的饱和CaO通过化学反应,生成Ca TiO3,Ca TiO3进一步还原成TiO,然后还原成金属Ti;随后K.Dring等[18]再次从热力学上证实了该还原过程中间产物存在的可能性.同时,C.Schwandt等[19]通过控制不同电压电解,检测到中间产物存在复合氧化物(Ca TiO3),且在低的电压下长时间电解TiO2得到产物金属Ti,更进一步确认了Ca析出电位前的TiO2的还原.
以上研究已经说明氧化物在熔盐中可以通过直接电化学还原途径得到金属.但是由于FFC法是在CaCl2的熔盐中进行,电解的过程中氧离子会从氧化物进入熔盐中,从而使熔盐中存在CaO.CaO的理论分解电压只有2.6 V(850℃),而TiO2的电解一般是在3.0~3.2 V的大电压下进行,因此阴极必然产生强还原能力的金属钙,这说明TiO2的还原过程同样存在钙热还原过程.
3.3 氧化物阴极还原动力学
对FFC工艺中氧化物阴极的还原过程,Chen等[20-21]建立了三相界线反应模型,认为反应首先发生在导电集流体∣固体氧化物∣熔盐三相接触区域,随着固体氧化物被还原为金属或导电的中间相,形成新的金属∣固体氧化物∣熔盐三相反应界线并不断扩展,从而使绝缘氧化物由表层到内层完成氧化物 到 金 属 的 转 变[22].T.Nohira 等[23-27]研 究 了SiO2在CaCl2熔盐中直接电化学还原,Jin等[28]在CaCl2熔盐中将多孔的二氧化硅电极直接还原得到硅的报道,认为SiO2的还原是由表层向里层发展的过程,SiO2的还原速度受O2-在阴极试片中的扩散速度控制.为了提高反应速度,阴极氧化物要制成疏松多孔的结构.但由于Si导电性能差,产物疏松多孔导致固相IR降明显,在产物深处Si∣SiO2∣熔盐界线上的极化电位甚至不足以使SiO2还原而导致反应停止.为此,T.Nohira等通过改进阴极氧化物结构使电解产物接触紧密,或在SiO2中掺Si等来提高导电性,减小固相IR降的影响,来提高反应速度.如果产物为导电性好的金属或合金,可以忽略固相产物IR降的影响,则可以直接通过改善阴极氧化物结构来加快反应速度.
3.4 对电解工艺条件的探索
在CaCl2熔盐进行电化学还原氧化物时,由于大多数的氧化物(如SiO2、TiO2、Zr O2)均为绝缘体或者导电性能不佳,还原过程将受到极化电位、氧离子的扩散以及氧化物颗粒与金属颗粒之间的欧姆电阻等因素的影响.基于对FFC方法的理论研究,研究者可以有针对性的探索最佳电解工艺条件,比如氧化物阴极结构优化、电解电压、时间以及反应温度的研究,从而提高电解的电流效率、降低能耗、提高电解产品的质量.蒋凯等发现现场钙钛矿化是FFC电化学还原二氧化钛不可避免的中间步骤,而这一步骤严重制约了其后续还原过程,导致整个电解过程速度较慢和电流效率较低.为了解决这一难题,研究者创新性地提出了非现场钙钛矿化的制备金属钛的新方法,发现在电解试验中,非现场钙钛矿化可显著提高电解速度和电流效率,从而为FFC法制备金属钛或其他材料的工业化生产提供了一条新的思路[4].
如上小节中所述,研究者普遍认为氧离子在多孔阴极试片中的扩散速度为还原的控制步骤,因此增大氧化物的孔隙率可以提高氧离子的扩散速度,从而提高氧化物的还原速度.通过在氧化物中添加造孔剂(碳粉、CaO等)来增大氧化物的孔隙率[29-30],也可以减少试片的压制压力或降低烧结温度等手段使氧化物的孔隙率提高[31].文献[32]通过研究TiO2在CaCl2熔盐中的电化学还原过程发现,TixO(x≥1)-Ti是还原过程的控制步骤,金属/金属氧化物摩尔体积比过大导致反应界面受到限制,进而归纳出该工艺提炼各种金属的电流效率与金属/金属氧化物摩尔体积比变化的规律.选择NH4HCO3作为造孔剂改善氧化物的孔隙率,找到适合该实验体系实现二氧化钛快速还原的最佳孔隙率范围(60%~70%),提出两步槽压电解法,得到了目前生产氧含量(质量分数)低于0.2%的金属Ti可以达到的最高的电流效率和最低的能耗.减少氧化物试片厚度也是提高氧离子的传质速度的有效手段[33].但是考虑到电解槽设计和产量,当氧化物阴极试片的厚度太小或者孔隙率极大时,氧化物的有效质量减少,从而得到的产物的量很少,这对生产效益是不利的.
3.5 FFC法的产业化进程
自FFC法报道以来,国内外的研究者对该方法进行了大量的研究.英国帝国理工学院著名专家Flower评论[34]指出,该工艺一旦实现工业化,将是钛生产的一个重大革命性变化和里程碑.因此工业界寄希望于用该方法替代传统的Kroll法进行钛等金属的冶炼.为了使剑桥大学研制成功的FFC工业得到商业化,英国在1998年组建了大不列颠钛业公司(British Titanium Corporation,BTi)并与剑桥大学及英国政府资助的DERA(Defense Evaluation and Research Agency)合作,对该技术进行孵化,开展公斤级规模的试验.2003年3月,美国DARPA投资1230万美元,资助由世界最大的金属钛生产企业Timet牵头的包括BTi、剑桥大学、加州伯克利分校等合作,计划在4年内分3步逐级放大实现熔盐电解固态TiO2工业化生产金属Ti,目前已实现公斤级的放大.澳大利亚的BHPBpition公司用与FFC和OS类似的方法(命名为Polar法),采用熔盐电解法生产金属钛,研究规模已经成功地从克级扩大到公斤级.2006年Metalysis公司获得了FFC法开发钛的权力,该公司结合FFC工艺和Polar工艺优点,生产钛及其合金.2010年,Metalysis和包括劳斯莱斯公司、波音集团、美国铝企巨头Alcoa等公司接洽,计划投资7000万英镑建设世界上首家低成本钛厂,如果该厂建成投产,钛价至少会降下一半[35].
目前,在这一领域的研究机构除了该工艺发源地英国剑桥大学,美国、澳大利亚、日本等国都有相关研究.国内在这方面也有众多研究机构参与,包括武汉大学、西北有研究院、北京有色金属研究总院、北京科技大学、中国过程工程研究所等.其中,武汉大学在这一领域的研究水平在国内乃至国际都处于领先地位.
4 结束语
以金属钛为代表的金属及合金在国民经济中具有重要地位,广泛应用于化工、冶金、船舶、汽车、航空航天工业、国防技术等领域.由于化石能源逐渐枯竭和二氧化碳排放对全球气候的影响等问题日趋严重,目前许多工业纯金属和合金的生产工艺被普遍认为需要改进,特别突出的问题是过程复杂、工艺流程长、能源消耗大、污染环境等,诸如此类问题还导致一些性能优良的金属、合金的价格居高不下,限制了其在各个领域的应用.作为具有革命性意义的一种材料制备和冶金新工艺,FFC工艺与常规工艺比较,是一项能使生产高价值合金和金属的厂家投资较小、对环境友好的更有吸引力的工艺.不过,FFC法的工业化研究还面临许多的困难,还有大量问题需要众多的研究者们去挑战和解决,如大规模生产所需电解池的细节设计、过程控制的改进、电解速度和电流效率如何提高、在电解池中阴极产物如何进行连续化生产等等.
[1]KROLL W.The production of ductile titanium [J].Tr Electrochem Soc,1940,78:35.
[2]CHEN G Z,FRAY D J,FARTHING T W.Direct electrochemical reduction of titanium dioxide to titanium in molten calcium chloride[J].Nature,2000,407:361.
[3]FRAY D J,FARTHING T W,CHEN G Z.Removal of oxygen from metal oxides and solid solutions by electrolysis in a fused salt:UK,WO9964638[P].1998-06-05.
[4]JIANG K,HU X H,MA M,et al.“Perovskitization”-assisted electrochemical reduction of solid TiO2in molten CaCl2[J].Angew Chem Int Ed,2006,45:428.
[5]SCHWANDT C,ALEXANDER D T L,FRAY D J.The electro-deoxidation of porous titanium dioxide precursors in molten calcium chloride under cathodic potential control[J].Electrochimica Acta,2009,54:3819-3829.
[6]STOHR U,FREYLAND W.Intervalence charge transfer and electronic transport in molten salts containing tantalum and niobium complexes of mixed valency[J].Phys Chem Chem Phys,1999,1:4383.
[7]ABEDIN S Z El,TERAKADO O,ENDRES F,et al.Inter-valence charge transfer in mixed valence neodymium iodide melts:electronic conductivity and optical absorption spectra[J].Phys Chem Chem Phys,2002,4:5335.
[8]肖巍.固态二氧化硅在氯化物熔盐中直接电化学还原过程及产物研究[D].武汉:武汉大学,2007.
[9]GAO P,JIN X,WANG D,et al.Preparation of a novel reference electrode in molten electrode[J].Electrochemistry(in Chinese),2005,11(1):8.
[10]GAO P,JIN X,WANG D,et al.A quartz sealed Ag/AgCl reference electrode for CaCl2based molten salts[J].J Electr Oanal Chem,2005,579:321-328.
[11]CHA C,LI C,YANG H,et al.Powder microelectrode[J].J Electroanal Chem,1994,368:47.
[12]JIANG K,HU X,SUN H,et al.Electrochemical synthesis of LiTiO2and LiTi2O4in molten LiCl[J].Chem Mater,2004,16:4324.
[13]QIU G,FENG X,LIU M,et al.Investigation on electro-chemical reduction process of Nb2O5powder in molten CaCl2with metallic cavity electrode[J].Electrochimica Acta,2008,53:4074-4081.
[14]SUZUKI R,INOUE S.Calciothermic reduction of titanium oxide in molten CaCl2[J].Metall Mater Trans B,2003,34(3):277-285.
[15]SUZUKI R,TERANUMA K,ONO K.Calciothermic reduction of titanium oxide and in-situ electrolysis in molten CaCl2[J].Metall Mater Trans B,2003,34(3):287-295.
[16]CHEN G Z,FRAY D J.Voltammetric studies of the oxygen-titanium binary system in molten calcium chloride[J].J Electrochem Soc,2002,149:E455-E467.
[17]DRING K,DASHWOOD R,INMAN D.Voltammetry of titanium dioxide in molten calcium chloride at 900℃[J].J Electrochem Soc,2005,152:E104.
[18]DRING K,DASHWOOD R,INMAN D.Predominance diagrams for electrochemical reduction of titanium oxides in molten CaCl2[J].J Electrochem Soc,2005,152:D184.
[19]SCHWANDT C,FRAY D J.Determination of the kinetic pathway in the electrochemical reduction of titanium dioxide in molten calcium chloride[J].Electrochim Acta,2005,51:66.
[20]CHEN G Z,FRAY D J.Novel cathodic processes in molten salts[C].Proceedings of the 6th International Symposium on Molten Salt Chemistry and Technology.Shanghai:Shanghai University Press,2001:79-85.
[21]CHEN G Z,FRAY D J.Understanding the electro-reduction of metal oxides in molten salts[J].Light Metals,2004:881-886.
[22]CHEN G Z,GORDO E,FRAY D J.Direct electrolytic preparation of chromium powder[J].Metall Mater Trans B,2004,35B:223.
[23]NOHIRA T,YASUDA K,ITO Y.Pinpoint and bulk electrochemical reduction of insulating silicon dioxide to silicon[J].Nature Materials,2003,2:397-401.
[24]YASUDA K,NOHIRA T,ITO Y.Effect of electrolysis potential on reduction of solid silicon dioxide in molten CaCl2[J].J Phys Chem Solids,2005,66:443.
[25]YASUDA K,NOHIRA T,AMEZAWA K,et al.Mech-anism of direct electrolytic of solid SiO2to Si in molten CaCl2[J].J Electrochem Soc,2005,152:D69.
[26]YASUDA K,NOHIRA T,TAKAHASHI K,et al.E-lectrolytic reduction of a powder-molded SiO2pellet in molten CaCl2and acceleration of reduction by Si addition to the pellet[J].J Electrochem Soc,2005,152:D232.
[27]YASUDA K,NOHIRA T,HAGIWARA R,et al.Direct electrolytic reduction of solid SiO2in molten CaCl2for the production of solar grade silicon[J].Electrochim Acta,2007,53:106-110.
[28]JIN X B,GAO P,WANG D H,et al.Electrochemical preparation of silicon and its alloys from solid oxides in molten calcium chloride[J].Angew Chem Int Edit,2004,43:733.
[29]邱国红.熔盐电化学还原固态氧化物制备重稀土金属铽及合金的基础研究[D].武汉:武汉大学,2006.
[30]CENTENO-SANCHEZ R L,FRAY D J,CHEN G Z.Study on the reduction of highly porous TiO2precursors and thin TiO2layers by the FFC-cambridge process[J].J Mater Sci,2007,42:7494.
[31]YAN X Y,FRAY D J.Electrochemical studies on reduction of solid Nb2O5in molten CaCl2-NaCl eutectic—I.Factors affecting electrodeoxidation of solid Nb2O5to niobium[J].J Electrochem Soc,2005,152:D12.
[32]LI W,JIN X B,HUANG F L,et al.Metal-to-oxide molar volume ratio:The overlooked barrier to solidstate electroreduction and a“green”bypass through recyclable NH4 HCO3[J].Angew Chem Int Ed,2010,49:3203-3206.
[33]XIAO W,JIN X B,DENG Y,et al.Electrochemically driven three-phase interlines into insulator compounds:Electroreduction of solid SiO2in molten CaCl2[J].Chem Phys Chem,2006,7:1750-1758.
[34]FLOWER H M.A moving oxygen story[J].Nature,2000,407:305.
[35]TAGARIS K.Metalysis seeks backer for titanium plant:report [EB/OL].London,2010-06-18.http://www.reuters.com/article/id USTRE66I0CW20100719.
Research development of preparation of titanium by the FFC cambridge process
LI Wei,WANG Ying,TANG Ren-heng,XIAO Fang-ming
(Department of Rare Metals,Guangzhou Research Institute of Non-ferrous Metals,Guangzhou 510650,China)
The basic principle of direct electro-reduction of solid TiO2to prepare Ti in molten salts is introduced,namely FFC-Cambridge process.The current research work and industrialization are also reviewed.The basic problems for FFC-Cambridge process industrialization are discussed in detail.At the end of this paper,we pointed out that FFC-Cambridge process is the most remarkable method of preparing titanium and its alloys,and the realization of industrialization can be expected in the near future.
FFC-cambridge process;TiO2;molten salts;Ti
O6
A
1673-9981(2010)04-0555-06
2010-10-25
李伟(1981—),男,湖南人,博士.
猜你喜欢
氯碱工业(2021年5期)2021-09-10
陶瓷学报(2020年6期)2021-01-26
湿法冶金(2020年1期)2020-02-24
陶瓷学报(2019年6期)2019-10-27
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
科学与财富(2017年9期)2017-06-09
中国氯碱(2017年3期)2017-04-18
电镀与环保(2016年3期)2017-01-20
材料科学与工程学报(2016年1期)2017-01-15
