
(1S,3S)-1,3-二苯基-1,3-丙二胺的合成*
2010-11-27 08:51:02陈垦,李杰,徐亮
合成化学 2010年3期
陈 垦, 李 杰, 徐 亮
(四川大学 华西药学院 天然药物学系,四川 成都 610041)
具有C2对称的手性1,3-二苯基-1,3-二胺(1)尽管在不对称催化反应中显示出潜在的应用价值[1],但相关合成方法的研究[2~5]却鲜见报道。Denmark 等[5]在1992年报道了以N-Boc保护的二氢吡唑(3)为原料,先将苯基锂与无水CeCl3交换形成苯基铈试剂(PhCeCl2),然后对亚胺键进行加成反应制得关键中间体四氢吡唑化合物4;4脱Boc保护后经催化氢化裂解氮氮键即得到消旋的1。该路线起始原料价廉易得,关键反应3→4立体选择性较高,但是反应所用试剂昂贵,反应条件十分苛刻,需要在高度无水无氧条件下进行。我们在重复文献[5]方法时发现该反应的可操作性和重复性都非常差,不易大量制备。
本文对文献[5]方法进行优化,改用价廉易得的苯基格氏试剂(PhMgBr)在六甲基磷酰胺(HMPA)存在下以二氯甲烷作溶剂与3反应,比较大量地制备了4; 再由4合成消旋的1(总收率35%);1经L-二苯甲酰酒石酸盐(L-DBTA)拆分[6]制得光学纯的(1S,3S)-1, 3-二苯基-1,3-丙二胺[(+)-1, Scheme 1]。
本方法具有成本廉价、操作简单、收率较高、可较大量制备等优点。
1 实验部分
1.1 仪器与试剂
Perkin-Elmer 341型旋光仪;Varian Unity-INOVA-400/54型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent 1200型系列高效液相色谱仪(CHIRALPAK AD-H手性柱)。
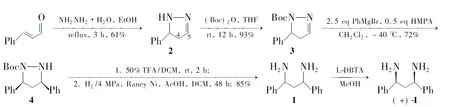
Scheme 1
柱层析用硅胶,200目~300目,青岛海洋化工厂;薄层层析GF254硅胶板,烟台江友硅胶开发有限公司。
1.2 合成[5]
(1) 3-苯基二氢吡唑(2)的合成
在三颈瓶中加入水合肼50 mL,搅拌下于室温缓慢滴加肉桂醛50 g(380 mmol)的乙醇(25 mL)溶液,滴毕,回流反应3 h。冷却至室温,分液,水层用乙酸乙酯(50 mL)萃取,合并有机层,用Na2SO4干燥,减压蒸干溶剂得黄色油状液体,减压蒸馏得无色油状液体2 33 g,收率61.2%;1H NMRδ: 7.41~7.26(m, 5H, PhH), 6.84(br s, 1H, 3-H), 4.73(t,J=9.6 Hz, 1H, 5-H), 3.15(dd,J=17.2 Hz, 11.6 Hz, 1H, 4-H), 2.72(dd,J=17.2 Hz, 8.4 Hz, 1H, 4-H)。
(2) 3的合成
氩气保护,在三颈中瓶加入2 33 g(230 mmol)的THF(100 mL)溶液,搅拌下于室温缓慢滴加(Boc)2O 58.5 g(260 mmol)的THF(50 mL)溶液,滴毕,反应过夜。蒸干溶剂,残留物经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=5 ∶1]分离得白色固体3 53 g,收率93%;1H NMRδ: 7.36~7.20(m, 5H, PhH), 6.91(br s, 1H, 3-H), 5.17(dd,J=12.1 Hz, 5.4 Hz, 1H, 5-H), 3.43(ddd,J=18.5 Hz, 12.1 Hz, 1.2 Hz, 1H, 4-H), 2.82(ddd,J=18.5 Hz, 5.4 Hz, 1.2 Hz, 1H, 4-H), 1.36(br s, 9H, CH3)。
(3) 4的合成
氩气保护,在反应瓶中依次加入3 19.68 g(80 mmol)的CH2Cl2(400 mL)溶液和HMPA 7.0 mL(40 mmol),搅拌下于-40 ℃缓慢滴加2.5 mol·L-1PhMgBr的Et2O溶液80 mL,滴毕,反应5 h。加入饱和NH4Cl水溶液(20 mL)和冰水(100 mL),充分搅拌,静置,分液,水层用CH2Cl2(3×70 mL)萃取,合并有机层,用饱和食盐水洗涤,无水Na2SO4干燥,减压蒸干,残余物经硅胶柱层析(洗脱剂:A=10 ∶1)分离得白色固体4 18.7 g,收率72%;1H NMRδ: 7.45~7.20(m, 10H, PhH), 4.89(t,J=6.8 Hz, 1H, 5-H), 4.76(br s, 1H, NH), 4.59(br s, 1H, 3-H), 2.76(m, 1H, 4-H), 2.40(m, 1H, 4-H), 1.32(br s, 9H, CH3)。
(4) 1的合成
氩气保护,在反应瓶中加入4 8 g(24.7 mmol)的CH2Cl2(17 mL)溶液,冰水浴冷却,搅拌下缓慢滴加三氟乙酸(TFA) 16.5 mL(215.6 mmol),滴毕,于室温反应2 h。减压蒸干,残余物用CH2Cl2(90 mL)转移至高压反应釜中,加入Raney-Ni 12 g和冰醋酸3.65 mL,在4 MPa下加压氢化反应40 h。用10%氨水充分碱化至pH>10,减压过滤,滤液分液,水层用CHCl3(4×20 mL)萃取,合并有机层,用饱和食盐水洗涤,无水Na2SO4干燥,蒸除溶剂得白色固体1 4.8 g,收率85%;1H NMRδ: 7.38~7.25(m, 10H, PhH), 3.94(t,J=6.8 Hz, 2H, 1,3-H), 2.05(t,J=6.8 Hz, 2H, 2-H)(与文献[5]值一致)。
1.3 1的拆分[6]
在烧杯中加入1 9.2 g和L-DBTA 14 g的甲醇(35 mL)溶液,搅拌下滴加乙醚2 mL,滴毕,静置12 h。过滤,滤饼用10%氨水充分碱化;滤液用CHCl3(3×15 mL)萃取,合并有机层,用饱和食盐水洗涤,无水Na2SO4干燥,减压蒸干溶剂。残余物再拆分一次;合并两次滤饼共得白色固体(+)-1 3 g, [α]D+33.0°(c0.1, 50%EtOH),ee>99%(HPLC)。
2 结果与讨论
由肉桂醛合成1的关键步骤是格氏试剂加成反应(3→4),本文重点讨论了反应条件对合成4的影响。
3 1 mmol, PhMgBr 2.5 mmol,反应温度-10 ℃,考察溶剂(5.0 mL)对合成4的影响,结果见表1。由表1可见,CH2Cl2为最佳溶剂。以CH2Cl2为溶剂,其余反应条件不变,考查反应温度对合成4的影响,结果见表2。从表2可以看出,反应温度降低至-40 ℃时,收率提高至70%;降低至-78 ℃时,收率略有降低。
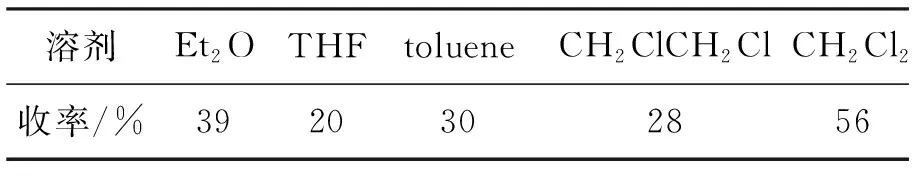
表 1 溶剂对加成反应的影响*Table 1 Effect of solvent on the addition reaction
*3 1 mmol, PhMgBr 2.5 mmol,反应温度-10 ℃,溶剂5.0 mL
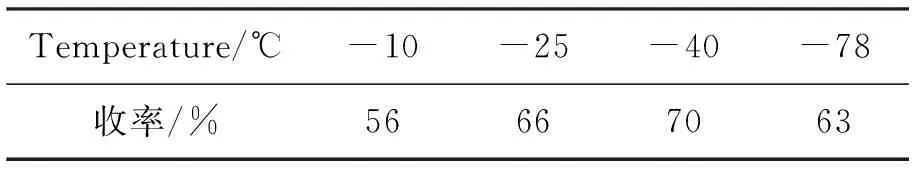
表 2 温度对加成反应的影响*Table 2 Effect of reaction temperature on the addition reaction
*以CH2Cl2为溶剂,其余反应条件同表1
由于HMPA[7]可以显著提高格氏试剂对亚胺加成反应的收率,因此,我们以CH2Cl2为溶剂,反应温度-40 ℃, 3 1 mmol, PhMgBr 2.5 mmol,考查了HMPA用量对加成反应的影响,结果见表3。由表3可见,HMPA能明显提高4的收率,其最佳用量为0.5 mmol,收率86%。
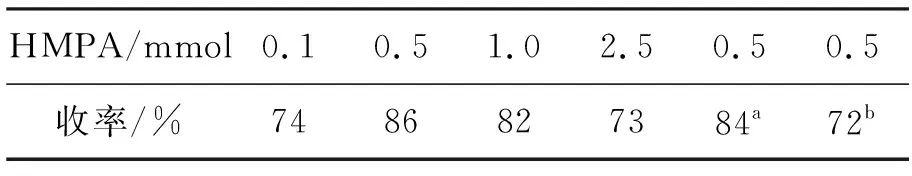
表 3 HMPA用量对加成反应的影响*Table 3 Effect of HMPA amount on the addition reaction
*3 1 mmol,n(3) ∶n(PhMgBr) ∶n(HMPA)=1.0 ∶2.5 ∶0.5,以CH2Cl2(5.0 mL)为溶剂,反应温度-40 ℃;a3 10 mmol;b3 80 mmol
综上所述,合成4的较适宜反应条件为: 3 1 mmol,n(3) ∶n(PhMgBr) ∶n(HMPA)=1.0 ∶2.5 ∶0.5,以CH2Cl2(5.0 mL)为溶剂,反应温度-40 ℃,收率86%。
在较适宜的反应条件下做放大实验(表3),3 10 mmol时,收率未明显降低(84%);3 80 mmol时,收率有所下降,但是仍然能够获得72%的较好收率。
[1] William P Hems, Michelle Groarke, Antonio Zanotti-Gerosa. [(Bisphosphine)Ru(Ⅱ) diamine] complexes in asymmetric hydrogenation:Expanding the scope of the diamine ligand[J].Acc Chem Res,2007,40:1340-1347.
[2] Gregory H P Roos, A Richard Donovan. Synthesis of novelC2-symmetric ligands based on (R,R)- and (S,S)-diphenyl-1,3-propanediol[J].Tetrahedron:Asymmetry,1999,10:991-1000.
[3] Gabriela A Grasa, Antonio Zanotti-Gerosa, William P Hems. A chiral [(dipyridylphosphine]RuCl2(1,3-diphenylpropanediamine)] catalyst for the hydrogenation of aromatic ketones[J].J Organmet Chem,2006,691:2332-2334.
[4] Alex Alexakis, Nathalie Lensen, Jean-Philippe Tranchier,etal. Reactivity and diastereoselectivity of Grignard reagents toward the hydrazone functionality in toluene solvent[J].J Org Chem,1992,17:4563-4565.
[5] Scott E Denmark, Jung-Ho Kim. A diastereoselective synthesis of (dl)-1,3-dDiphenyl-1,3-propanediamines[J].Synthesis,1992:229-234.
[6] Sumio Arakawa, Kazuo Kashiwabara, Junnosuke Fujita,etal. Preparation of optically active 1,3-diphenyl-1,3-propanediamines(dppn) and the circular dichroism oftrans-[CoCl2-(S,S-dppn)2]+and [Co(S,S-dppn)3]3+[J].Bull Chem Soc Jpn,1977:2108-2112.
[7] A I Meyers, Eric W Collington. Synthesis via-oxazolines.Ⅰ.The formylation of Grignard reagents in the presence of hexamethylphosphoramide[J].J Am Chem Soc,1970,92:6676-6678.
猜你喜欢
医学信息(2024年23期)2024-12-12 00:00:00
江南诗(2023年6期)2023-12-08 05:17:24
金沙江文艺(2022年1期)2022-02-04 10:15:16
今日农业(2021年2期)2021-11-27 19:19:53
化学教与学(2021年12期)2021-02-18 01:16:58
今日农业(2020年23期)2020-12-31 09:00:40
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24 21:02:17
中成药(2017年10期)2017-11-16 00:50:15
中成药(2017年4期)2017-05-17 06:09:46
合成化学(2015年1期)2016-01-17 08:59:30
