
叠氮化氰的光解离机理
2010-11-06 07:01慈成刚段雪梅刘靖尧孙家钟
物理化学学报 2010年10期
慈成刚 段雪梅 刘靖尧 孙家钟
(吉林大学理论化学研究所,理论化学计算国家重点实验室,长春 130023)
叠氮化氰的光解离机理
慈成刚 段雪梅 刘靖尧*孙家钟
(吉林大学理论化学研究所,理论化学计算国家重点实验室,长春 130023)
采用多参考态方法,在MRCI+Q//CAS(10,9)/6-311+G(2df)水平上对叠氮化氰(N3CN)的光解离机理进行理论研究.优化得到基态(S0)和低激发态(S1、S2、T1)势能面上的极小点、过渡态、内转换交叉点(IC-S1/S0)和隙间窜跃交叉点(ISC-S1/T1)的结构和能量,构建反应势能面.在MRCI+Q//CAS(10,9)水平上计算N3CN的垂直激发能,并和实验值进行对比.结果表明,在S0、S1、S2和T1态势能面上,N—N键断裂生成N2+NCN是主要解离途径,而C—N键断裂通道是次要通道.实验观测到220 nm处的吸收峰对应分子由S0态到S1态的激发,对应主要光解离产物为NCN[a1△g];而在275 nm处的吸收峰则对应分子被激发到T1态,然后直接生成基态产物NCN[X3Σ-g].我们的理论结果与实验测量符合得很好.
反应机理; 叠氮化氰; 光解离; 势能面交叉点
叠氮化氰(N3CN)作为叠氮化物[1-4]的氰基取代物,是合成化学中一种有用的高活性试剂[5].早期的一些实验[6-10]研究了它的光解离过程.实验发现, N3CN在紫外光下可解离生成NCN,CN,N3等自由基.这些自由基由于具有重要的性质,近年来受到了实验和理论的广泛关注,如双自由基NCN,既是燃烧化学中的重要中间体[11],也是合成高硬度碳氮材料的优良增长物种[12].因此,鉴于其光解产物的重要性,从理论上详细了解N3CN的光解离机理是十分必要的.
Marsh和 Hermes[13]在环己烷溶液中观测到N3CN有两个主要吸收带,最大峰值分别在275和220 nm处,其中220 nm处为强吸收峰,摩尔消光系数比275 nm处的强约20倍.Kroto等[6]在对N3CN的光解实验研究中,提出N3CN存在两种光解离途径:

并且认为通道(1)为主要解离通道,对应解离产物为基态N2和NCN(1△g),通道(2)为次要光解离通道.Okabe和Mele[7]用短波长(λ≤206.2 nm)光源强行光解离N3CN,得到了与Kroto等[6]相同的结果.Milligen等[8-9]和Shoen[10]通过基质分离技术研究了N3CN的光解离过程,提出了与Kroto等[6]略有不同的机理,指出N3CN在波长275 nm处先跃迁到一个不稳定的三重态,然后解离成NCN(3Σ-g)+N2;而在波长220 nm处伴随着一个到不稳定单重态的跃迁,然后解离生成NCN(1△g)+N2.
理论上,Jensen[14]和 Türker等[15]分别计算了N3CN的基态几何结构,他们的计算结果和实验数据[16-17]符合得很好.Benard等[12]利用B3LYP/6-311+ G(2df)方法,在Cs对称性下,对N3CN分子中的NN—NCN键做了扫描,得到了1A′和3A″态下的扫描势能曲线.结果表明在3A″态下,NN—NCN键断裂是一个直接解离过程,并且发现3A″态的势能曲线末端与1A′态势能曲线存在势能面交叉.但他们的计算中没有给出解离机理以及势能面交叉点的信息.因此,本文将通过多参考态量子化学计算方法,研究N3CN基态和几个低激发态稳定点的结构,构建反应势能面,进而确定叠氮化氰的光解离机理.
1 计算方法
采用全活空间自洽场方法(CASSCF)[18-19]对N3CN基态和激发态稳定点的几何结构进行优化,并通过频率分析以确定得到的稳定点是势能面上的极小点(0个虚频)或过渡态(1个虚频).为了确定过渡态两边正确的连接关系,在CASSCF水平上进行了内禀反应坐标(IRC)[20]计算.使用态平均全活化空间自洽场方法(SA-CASSCF)确定势能面交叉点的结构.另外,为了包含动态相关能,在CASSCF优化结构基础上,应用多参考组态相互作用 (MRCI)[21-22]方法,同时考虑Davidson校正(MRCI+Q),进行单点能校正以得到体系更精确的能量值.
活化空间的选取是CASSCF计算的重要步骤.在此我们选取10个电子9个轨道的活化空间进行计算,记作CAS(10,9).活化空间包括:N1═N3键的面上π和π*轨道,N3≡N4键的面上π和π*轨道及C2≡N5键的面内π成键轨道.此外,因所研究体系涉及C2—N1和N1═N3键的断裂,所以C2—N1键的σ和σ*轨道及N1═N3键的σ和σ*轨道也被包含在活化空间内.在MRCI+Q计算中,以CASSSF计算结果作为参考波函数,C和N原子的1s电子作为冻结核轨道.文章中的计算基组全部采用6-311+G(2df).所有的计算均采用Molpro2006.1版本的程序包[23]完成.
2 结果与讨论
2.1 平衡几何结构和能量
图1给出了在CAS(10,9)/6-311+G(2df)水平上优化得到的基态N3CN(S0-MIN)的平衡几何结构(原子序号、键长和键角标于图中),作为对比将实验[16-17]及前人的理论计算[15]结果同时列于图中.可以看出本文的计算结果与已有的实验和理论结果符合得很好.基态N3CN为Cs对称性,其中N1—C2的键长是0.1351 nm,明显短于CH3HN2中的C—N单键键长0.1470 nm[24],这表明N1═N3键和C2≡N5键间存在很强的共轭作用.
在CAS(10,9)/6-311+G(2df)水平上,优化得到了S1,T1和S2态的稳定点,分别记作S1-MIN,T1-MIN和S2-MIN,构型见图1.S1-MIN具有Cs对称性,构型改变主要集中在N1—N3—N4区域.可以看出π→π*跃迁导致S1态的N1—N3键长由基态的0.1270 nm增长到0.1410 nm,N3—N4键比基态的伸长了0.0075 nm,同时,N1—N3—N4键角由基态的171.9°减小到117.7°,而N1—C2—N5区域的键长和键角与基态相比几乎不变.T1态的稳定点结构与S1态类似,它们之间的最大差别在于T1-MIN不具有Cs对称性,相应的二面角N4—N3—N1—C2为116.0°,即N3N4部分扭转出分子平面.分子激发到S2态后,基本上保持一个平面结构.和基态稳定点相比,构型改变主要集中在N1—C2—N5区域.可以看出N5≡C2键伸长了0.0109 nm,N1—C2键缩短了0.0091 nm.

基于基态稳定构型,在MRCI+Q/6-311+G(2df)水平上对 N3CN的垂直激发能进行了计算,在Frank-Condon区域按能量由低到高的次序依次为13A″、11A″和 21A′,分别对应 T1、S1和 S2态.在MRCI+Q水平上计算得到的11A′→13A″跃迁的垂直激发能为476.0 kJ·mol-1.结合实验所测光谱数据可知,对应吸收峰为275 nm(435.1 kJ·mol-1)处的跃迁.MRCI+Q计算的结果略高于实验值.而实验上220 nm(543.9 kJ·mol-1)处吸收峰则对应11A′→11A″的跃迁.理论计算的垂直激发能为506.5 kJ·mol-1,略低于实验值.通过自然轨道分析可知11A′→13A″和11A′→11A″的跃迁均为电子由N1—N3—N4面上π轨道向π*轨道的激发.此外,在MRCI+Q//CAS (10,9)/6-311+G(2df)水平上,S0态到T1、S1和S2态的绝热激发能分别为249.0、232.3和499.9 kJ·mol-1.
势能面之间的交叉点在光解离反应中扮演着一个重要的角色,使用SA-CAS(10,9)/6-311G+(2df)方法寻找势能面之间的交叉点,在Frank-Condon区域,我们找到S1态和S0态之间及S1态和T1态之间的势能面交叉点,分别记作IC-S1/S0和ISC-S1/ T1.IC-S1/S0是一个N3—N4键扭转远离C2—N5键的构型,而ISC-S1/T1则是一个N3—N4键靠近C2—N5键的接近平面构型.在MRCI+Q水平上, IC-S1/S0和ISC-S1/T1的能量分别比S1态稳定点的高44.6和6.4 kJ·mol-1.
2.2 S0、T1、S1和S2态上N1═N3键的断裂
图2绘出了在MRCI+Q水平上,N3CN在S0, S1,T1和S2态的反应势能面示意图.S0态上N3CN的N1═N3键断裂过程存在一个过渡态TSN—N(S0),此时,N1—N3键伸长到0.1783 nm,C2—N1—N3键角减小到101.9°,N1—N3—N4和N1—C2—N5角度基本没发生改变.MRCI+Q计算得到的反应能垒为146.2 kJ·mol-1.IRC计算表明此过渡态在反应物方向连接着S0稳定点,产物方向连接着NCN [a1△g]+N2[X1Σ+g]碎片,相对能量为53.0 kJ·mol-1.
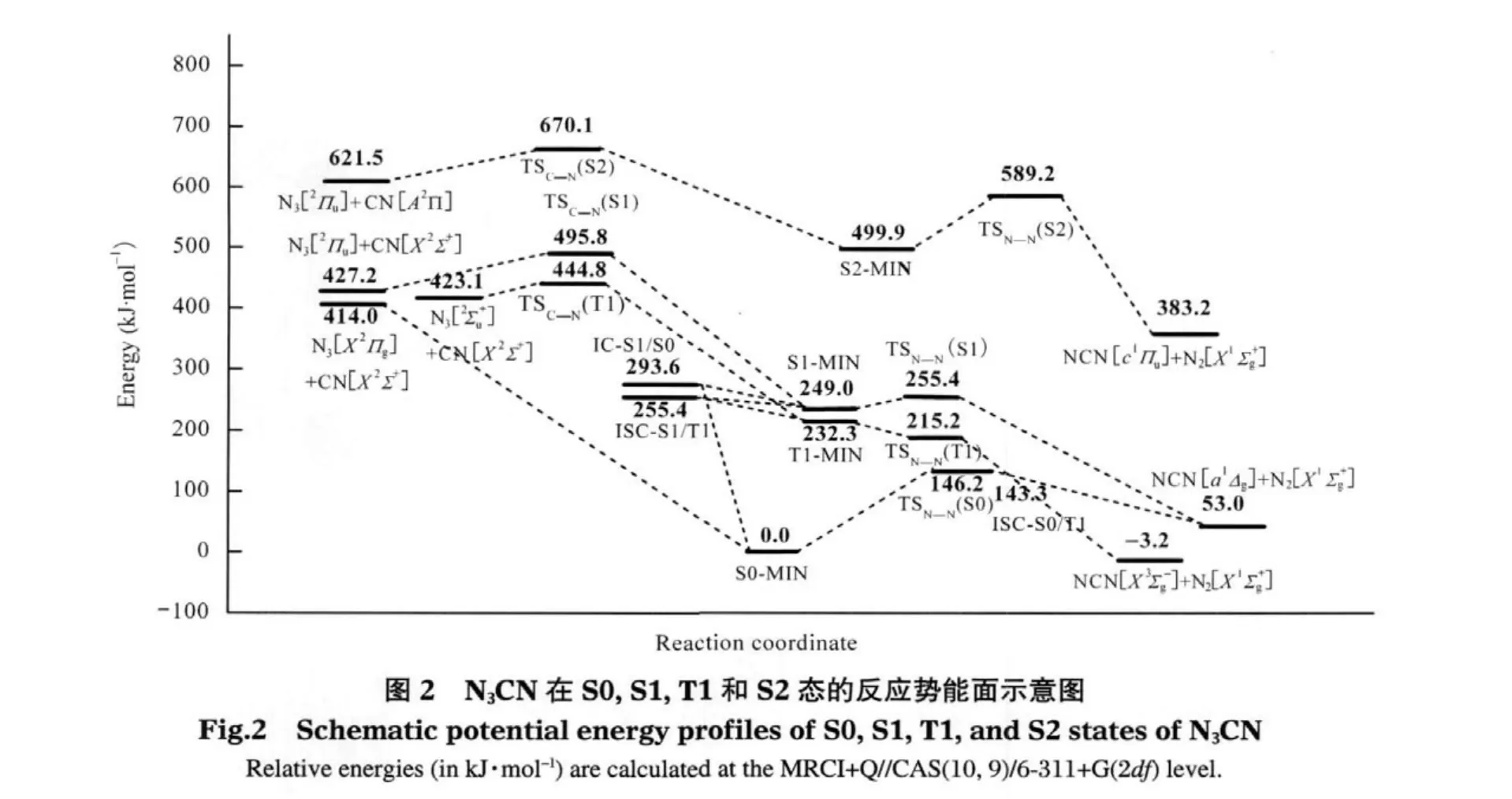
在CAS(10,9)/6-311+G(2df)水平上,优化得到S1态上N1—N3键断裂的过渡态TSN—N(S1),其中N1—N3键伸长至0.1622 nm,频率分析显示唯一的虚振动模式为N1—N3的伸缩振动.经过MRCI+Q高水平单点能校正后,TSN—N(S1)的能量仅比 S1-MIN高6.4 kJ·mol-1.在S1态,N1—N3键断裂是一个很容易发生的过程.IRC计算显示TSN—N(S1)连接S1态稳定点和S0态产物NCN[a1△g]+N2[X1Σ+g].
TSN—N(T1)为CASSCF水平上T1态N1—N3键解离的过渡态,连接三重态基态产物NCN[X3Σ-g]和N2[X1Σ+g]分子.此解离通道在CASSCF水平上存在反应能垒(13.8 kJ·mol-1).然而,经过MRCI+Q水平的单点能量校正后,该过渡态的相对能量为215.2 kJ·mol-1,比T1-MIN(232.3 kJ·mol-1)反应物的还低17.1 kJ·mol-1.鉴于此情况,我们对反应路径上的点都进行MRCI+Q单点能量计算,结果表明在T1态, N1—N3键的断裂过程无反应能垒存在,是一个直接解离的放热过程,也就是说,TSN-N(T1)并不是一个真正的反应过渡态.这个结果与Benard等[12]用B3LYP/6-311+G(2df)方法,证明3A″态是一个排斥态的结论相符.MRCI+Q计算得到的NCN[X3Σ-g]+N2[X1Σ+g]的相对能量为-3.2 kJ·mol-1.
另外,利用SA-CAS(10,9)/6-311+G(2df)方法在靠近基态N1═N3键解离产物方向找到了S0态与T1态势能面的交叉点(ISC-T1/S0),意味着N3CN分子在S0态的解离可以通过此交叉点生成T1态的基态产物.ISC-T1/S0的结构中除了N1—N3键长伸长至0.1820 nm外,其它的键参数和S0态的过渡态TSN—N(S0)十分接近.在MRCI+Q水平上,此交叉点低于S0态过渡态2.9 kJ·mol-1.Benard等[12]曾在B3LYP/6-311+G(2df)水平上,以基态N3CN构型为基础,扫描得到了3A″态和1A′态上N1—N3解离势能曲线,发现在N1—N3键长约为0.18 nm处存在一个势能面交叉点.可以看到他们的计算结果和本文的相一致.
在S2态势能面上同样找到了N1—N3键解离的过渡态,TSN—N(S2),相应的产物碎片为NCN[c1Πu]+ N2[X1Σ+g].此时N1—N3键长为0.1554 nm,比相应的S2-MIN伸长了 0.0210 nm,N1—N3—N4键角由S2-MIN中的125.1°变为115.6°.在MRCI+Q水平上,此反应能垒为89.3 kJ·mol-1.
2.3 S0、S2、S1和T1态上的N1—C2键断裂
在S0态势能面上,没有找到N3CN的N—C键断裂的过渡态,我们发现此反应是直接键解离的吸热过程,对应的解离产物为N3[X2Πg]+CN[X2Σ+].在MRCI+Q水平上,N—C键解离能为414.0 kJ·mol-1.在298 K,此通道的反应焓为393.8 kJ·mol-1,可以看到MRCI+Q的结果和实验值(399.3±20)kJ·mol-1(其中N3CN,(450±20)kJ·mol-1;CN,435.1 kJ·mol-1;N3, 414.2 kJ·mol-1)[25]符合得很好.
从S1和S2态的稳定点出发,在CAS(10,9)/6-311+G(2df)水平上,分别找到了N1—C2键断裂的过渡态TSC—N(S1)和TSC—N(S2),对应的解离产物分别为N3[2Πu]+CN[X2Σ+]和N3[2Πu]+CN[A2Π].与S1和S2态上各自的极小点相比,这两个过渡态的构型中:N1—C2键明显伸长,而N1—N3键的距离变短,接近正常N═N双键键长,同时N1—C2—N5键角分别由各自极小点的接近180°扭曲成153.5°和109.4°.另外,在TSC—N(S2)中,C2≡N5键扭曲伸出分子平面,几乎与N1—N3—N4面垂直,二面角N5—C2—N1—N3由S2-MIN中的180°变为-98.4°.在MRCI+Q//CAS(10,9)水平上,S2态和S1态的C2—N1键的活化能垒分别为170.2和246.8 kJ· mol-1.在T1态,我们虽然也找到了C—N键解离的过渡态,然而由于在T1态势能面沿着N—N键解离是一个排斥态,因此,认为此反应路径并不具有实际意义.
值得注意的是,对此体系,我们曾在CASPT2/6-311+G(2df)水平上进行了单点能量校正.但计算结果表明,在CASPT2//CASSCF水平上得到的垂直激发能以及基态N3CN的N—N键和C—N键的解离能都与实验值存在较大误差,也就是说,CASPT2方法不足以正确描述此反应体系的能量性质.因此,对于本文研究体系,需要使用更加精确的MRCI方法进行计算.
2.4 N3CN的光解离机理
由以上计算可以看到,N3CN分子存在两条光解离途径,即N—N键和C—N键的解离通道.由于在S0,S1,S2和T1势能面,前者的反应能垒远远低于后者(约80.9 kJ·mol-1或以上),因此,N—N键的断裂(反应通道(1))是N3CN分子在基态和三个低激发态势能面上绝对主要的解离通道,而C—N键解离是次要反应途径,只可能在一定的激发能下观测到,理论上预测的光解离机理和实验推测[6-7]相一致.
另外,实验上观测到N3CN分子的光解在220和275 nm处有两个吸收峰[5].计算表明220 nm处的吸收峰对应分子由S0到S1态的激发.当分子被激发到S1-MIN后主要存在三种去活化方式:一是经由过渡态TSN—N(S1)解离生成NCN[a1△g]+N2[X1Σ+g],即
S1-MIN→TSN—N(S1)→NCN[a1△g]+N2[X1Σ+g]
这个过程需要克服6.4 kJ·mol-1的能垒;二是经由交叉点IC-S1/S0内转换到S0态,再经过TSN—N(S0),生成同上的解离产物碎片,或在S0态TSN—N(S0)的解离产物路径方向,经过隙间窜跃交叉点 ISCS0/T1生成基态产物NCN[X3Σ-g]+N2[X1Σ+g].具体解离途径可表示成:

这个过程中,交叉点IC-S1/S0的能量比S1-MIN高出44.6 kJ·mol-1;三是由S1态经由交叉点ISC-S1/ T1隙间窜跃到T1态,继而发生无能垒的N—N键解离生成基态产物NCN[X3Σ-g]+N2[X1Σ+g].此交叉点能量比S1-MIN高出6.4 kJ·mol-1.此解离途径可以表达成:

对比这三种反应通道,可知N3CN分子在S1态上的绝热解离是最可能的去活化途径.此外,在275 nm处的吸收峰相应于N3CN分子从S0态到T1态的跃迁,由于这是个自旋禁阻的过程,因而实验上观测到这个吸收峰强度很弱.分子激发到T1态后,从T1态Frank-Condon点开始驰豫,沿着N—N键解离反应路径无能垒地生成基态产物NCN[X3Σ-g]+N2[X1Σ+g].本文在MRCI+Q//CAS(10,9)水平上得到的N3CN光解离机理与实验推测[8-10]的反应机理一致.
3 结 论
在MRCI+Q//CAS(10,9)/6-311+G(2df)水平上,计算了N3CN分子在基态和低激发态(S1、S2和T1)上的解离势能面.研究了其光解离机理并分析了可能的解离产物.结果表明,在S0、S1、S2和T1态上, N—N键断裂是N3CN绝对主要的解离通道,而C—N键断裂是次要解离途径.并且通过垂直激发能计算可以确定实验上观测到的275和220 nm处吸收峰分别对应S0→T1和S0→S1态的跃迁.在S1态上,N3CN可以经过IC-S1/S0交叉点发生内转换到S0态或通过ISC-S1/T1交叉点发生隙间窜跃到T1态,但在S1态上绝热解离生成NCN[a1△g]是最有利的反应途径.在T1态上,N3CN则沿着N—N键断裂反应路径无能垒地解离生成基态产物NCN[X3Σ-g].本文理论计算得到的光解离机理与实验推测一致.
1 Xiao,H.M.;Li,Y.F.;Qian,J.J.Acta Phys.-Chim.Sin.,1994,10 (3):235 [肖鹤鸣,李永富,钱建军.物理化学学报,1994,10(3): 235]
2 Xu,W.Y.;Liu,G.S.;Peng,Y.Y.;Hong,S.G.Acta Phys.-Chim. Sin.,1998,14(7):669 [徐文渊,刘够生,彭以元,洪三国.物理化学学报,1998,14(7):669]
3 Li,J.S.;Xiao,H.M.Acta Phys.-Chim.Sin.,2000,16(1):36 [李金山,肖鹤鸣.物理化学学报,2000,16(1):36]
4 Javad,H.;Naader,A.;Soraia,M.;Mehdi,A.Acta Phys.-Chim. Sin.,2009,25(6):1239 [Javad,H.;Naader,A.;Soraia,M.; Mehdi,A.物理化学学报,2009,25(6):1239]
5 Marsh,F.D.J.Org.Chem.,1972,37:2966
6 Kroto,H.W.J.Chem.Phys.,1965,44:831
7 Okabe,H.;Mele,A.J.Chem.Phys.,1969,51:2100
8 Milligen,D.E.;Jacox,M.E.;Bass,A.M.J.Chem.Phys.,1965, 43:3149
9 Milligen,D.E.;Jacox,M.E.J.Chem.Phys.,1965,45:1387
10 Schoen,L.J.J.Chem.Phys.,1965,45:2773
11 Jennings,K.R.;Linnett,J.W.Faraday Soc.,1960,56:1737
12 Benard,D.J.;Linnen,C.;Harker,A.;Michels,H.H.;Addision,J. B.;Ondercin,R.J.Phys.Chem.B,1998,102:6010
13 Marsh,F.D.;Hermes,M.E.J.Am.Chem.Soc.,1964,86:4506
14 Jensen,J.O.J.Mol.Struct.-Theochem,2005,730:235
15 Türker,L.;Atalar,T.J.Hazard.Mater.,2008,153:966
16 Costain,C.C.;Kroto,H.W.Can.J.Phys.,1972,50:1453
17 Almenningen,A.;Bak,B.;Jansen,P.;Strand,T.G.Acta Chim. Scand.,1973,27:1531
18 Werner,H.J.;Knowles,P.J.J.Chem.Phys.,1985,82:5053
19 Knowles,P.J.;Werner,H.J.Chem.Phys.Lett.,1985,115:259
20 Eckert,F.;Werner,H.J.Theor.Chem.Acc.,1998,100:21
21 Werner,H.J.;Knowles,P.J.Chem.Phys.,1988,89:5803
22 Knowles,P.J.;Werner,H.J.Chem.Phys.Lett.,1988,145:514
23 Werner,H.J.;Knowles,P.J.;Lindh,R.;et al.MOLPRO,a package of ab initio programs.version 2006.1
24 Butler,G.B.;Berlin,K.D.Foundation of organic chemistry (theory and application).Trans.Zhang,L.P.;Tu,Y.R.Beijing: People Education Press,1980:501 [Butler,G.B.;Berlin,K.D.有机化学基础(理论和应用).张丽蘋,涂余如,译.北京:人民教育出版社,1980:501]
25 NIST Chemisty Webbook[DB].Linstrom P.J.;Mallard W.G. Eds.Available from:http://webbook.NIST.Gov/chemistry
Photodissociation Mechanism of Cyanogen Azide
CI Cheng-Gang DUAN Xue-Mei LIU Jing-Yao*SUN Chia-Chung
(State Key Laboratory of Theoretical and Computational Chemistry,Institute of Theoretical Chemistry, Jilin University,Changchun 130023,P.R.China)
We investigated the photodissociation mechanism of cyanogen azide(N3CN)at the MRCI+Q//CAS(10, 9)/6-311+G(2df)level of theory using the multi-reference state method.The optimized structures and energies of the minima,transition states,singlet/singlet conical intersection and singlet/triplet crossing points of the ground and lowlying excited states were obtained to explore the potential energy surfaces of N3CN.The vertical excited energies calculated at the MRCI+Q//CAS(10,9)level were compared with the experimental data.It is shown that N—N bond fission to form N2+NCN is the predominant dissociation pathway on the S0,S1,S2,and T1 surfaces whereas the C—N bond fission channel is the minor pathway.The 220 nm absorption peak observed experimentally corresponds to an excitation from the S0 to the S1 state leading to the major photodissociation product NCN[a1△g].The 275 nm absorption peak corresponds to the S0-T1 transition leading to the formed ground-state product NCN[X3Σ-g]via the barrierlessly direct dissociation in the T1 state.Our theoretical results agree well with experimental observations.
Reaction mechanism; Cyanogens azide; Photodissociation; Potential energy surface intersection point
O641
Received:May 4,2010;Revised:June 2,2010;Published on Web:July 13,2010.
*Corresponding author.Email:ljy121@jlu.edu.cn;Tel:+86-431-8498016;Fax:+86-431-8498026.
The project was supported by the National Natural Science Foundation of China(20333050,20303007,20973077)and Program for New Century Excellent Talents in University,China(NCET).
国家自然科学基金(20333050,20303007,20973077)和教育部新世纪优秀人才支持计划(NCET)资助项目
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
吉林师范大学学报(自然科学版)(2022年4期)2022-12-09
北京航空航天大学学报(2022年5期)2022-06-06
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
World Journal of Psychiatry(2020年4期)2020-07-11
小学生学习指导(中年级)(2019年3期)2019-04-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
