
系统性红斑狼疮继发嗜血细胞综合征1例
2010-10-20 05:52陈柏叡仲少敏倪春雅李若瑜
实用皮肤病学杂志 2010年4期
陈柏叡,郭 静,仲少敏,倪春雅,赵 邑,李若瑜
临床资料
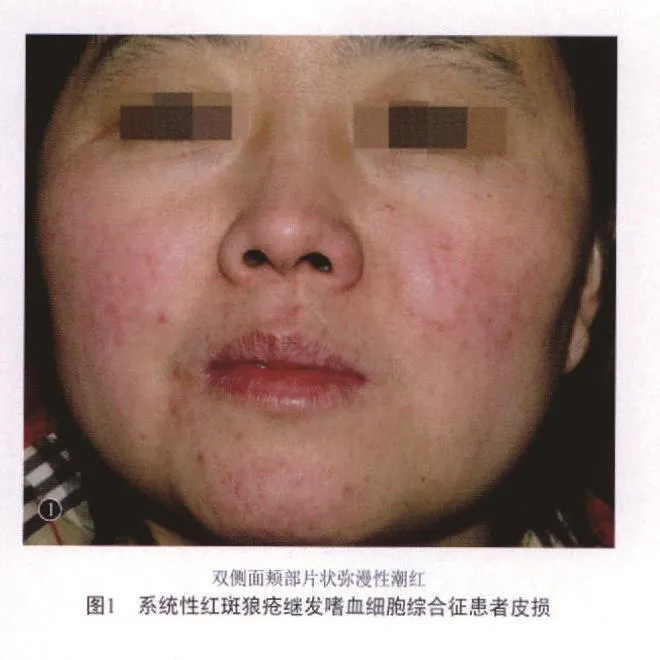
患者,女,27岁。主因面部皮疹2年,伴发热1周,于2008年9月12日来我科就诊。患者2年前无明显诱因左下眼睑出现红色斑疹,无痛痒,面积渐增大;2个月后,右面颊部又出现一硬币大小暗红斑,表面鳞屑,伴全身乏力,否认关节痛、腹痛、口腔溃疡及光敏史等,遂于我科门诊查抗核(ANA)抗体1∶1280,抗双链DNA(ds-DNA)抗体(-),抗nRNP(+),抗 Sm(-),尿蛋白(-),血常规 WBC 2.88×109/L(4~10×109/L),PLT 91×109/L(100~300×109/L),红细胞沉降率(ESR)30mm/h(15~20mm/h),诊断为“系统性红斑狼疮”。予强的松片30mg口服1次/d、羟氯喹片0.2g口服1次/d,长期服用并缓慢减量,1年前强的松片减至10mg口服1次/d,左下眼睑皮疹消退,期间复查白细胞始终低于4×109/L。本次发病为1个月前,患者自觉乏力、咽部异物感,复查血清补体C3 2.3mg/dl(60~150mg/dl),C4 0.1mg/dl(12~36mg/dl),IgA 426g/L(0.76~ 3.9g/L),ds-DNA 377 IR/ml(0~ 100 IR/ml),WBC 2.41×109/L,未予特殊处理;1周前出现发热,体温38.5℃~39.5℃,遂将强的松片改为强的松龙片60mg口服1次/d,羟氯喹片加量至0.2g口服2次/d,症状无缓解。体格检查:未触及浅表淋巴结肿大,心肺腹未见异常。皮肤科情况:双面颊部、双手掌片状弥漫性潮红(图1),表面无鳞屑,双手掌指腹部散在粟粒大小红斑,无鳞屑。辅助检查示:WBC1.88×109/L,EO% 0(0.01~ 0.05%),NE 1.24×109/L(1.4~ 6.5×109/L),LY 0.52×109/L(1.0~ 3.3×109/L),ESR 55mm/h(0~20 mm/h),ALT 61IU/L(0~ 40 IU/L),AST 62IU/L(0~ 40 IU/L),LDH 381IU/L(119~229 IU/L),HBDH 353IU/L(90~250IU/L);转铁蛋白2.25 mg/L(0~1.9 mg/L),血清铁蛋白>1500 ng/ml(10.0~292.0 ng/ml),总铁结合力 80.91 μmol/L(54~77 μmol/L),血清铁 4.7 μmol/L(50~ 175μmol/L);ANA 阳性(颗粒型)1∶3200,nRNP(+),Sm(+),SS-A(+),ENA 谱(-),IgA 4.39 g/L,补体C3 0.30g/L,补体C4 0.08g/L;尿蛋白(++),隐血或红细胞微量,高倍镜下红细胞4个;胸片检查未见异常。诊断为系统性红斑狼疮活动期。予甲强龙片250mg/d静点5日治疗,同时每日静点丙种球蛋白10g及重组人粒细胞集落刺激因子150mg,仍反复发热。因患者血象白细胞、红细胞及各细胞分类计数均明显减少,考虑存在嗜血现象,查巨细胞病毒 DNA(CMV-DNA) 和 EB 病 毒 DNA(EBV-DNA)均示阴性,多次血培养结果未见细菌感染,行骨髓穿刺检查示WBC、PLT减少,骨髓增生不良,易见嗜血细胞(图2a-2c),未见巨核细胞。为预防患者感染予以隔离并阿昔洛韦注射液250mg静点1次/d,将甲强龙改为强的松龙片25mg口服2次/d及利血生片20mg口服3次/d,反复输注新鲜血小板。经治疗,患者发热症状于约3日后逐渐下降至正常体温,复查血常规恢复至WBC 7.07×109/L,最后诊断为“系统性红斑狼疮继发嗜血细胞综合征”。给予阿莫西林胶囊0.4g口服2次/d,甲强龙片40mg口服2次/d,环孢霉素片350mg口服1次/d,持续治疗半个月后,患者出现面部、四肢水肿,皮肤多发瘀血瘀斑,口腔多发溃疡,并出现血压升高、血糖及转氨酶增高、消化道不适等药物性损害,血常规三系减低,血浆半乳甘露聚糖检测(GM试验)明确诊断为肺部烟曲霉感染。给予伏立康唑片400mg口服1次/d,并调整泼尼松、环孢素A及各项保护胃粘膜、补钾、钙剂等药物治疗,感染症状有所缓,激素逐渐减量至甲强龙片口服20mg/d,环孢霉素片100mg/d,出院时,病情控制稳定。嘱患者定期复查感染、肝功、电解质及血糖情况,但患者转诊于外院后失访。

讨论
嗜血细胞综合征(hemophagocytic syndrome,HPS)分成原发性(家族性)和继发性两类[1]。家族性嗜血细胞综合征(family hemophagocytic syndrome,FHS)是一种少见的常染色体隐性遗传病。患儿出生时正常,多于6~12个月时突然高热、黄疸、出血、肝脾肿大,实验室检查全血细胞减少、肝功能受损、凝血酶原时间延长、白蛋白降低、骨髓增生活跃、嗜血组织细胞增多。另一类是由肿瘤或感染所致的继发性嗜血细胞综合征,其又可分为恶性肿瘤相关性嗜血细胞综合征(maligant associated hemophagocytic syndrome,MAHS)和感染相关性嗜血细胞综合征(infection associated hemophagocytic syndrome,IAHS)两种。与SLE相关的HPS[2]属于继发性嗜血细胞综合征,分为①狼疮急性活动时并发的HPS;②免疫抑制剂治疗下无狼疮活动、与感染相关的HPS。
IAHS发病年龄多较早[3],从新生儿至18岁发病,50%在3岁以内,28%于1岁以内,新生儿极少见。病原有病毒、细菌和真菌,亦有支原体、立克次体、结核杆菌、杜氏利什曼原虫等。病毒以EB病毒及微小病毒B19最常见。嗜血细胞综合征的诊断标准[4]:①发热超过1周,体温≥38.5°C;②肝脾肿大,伴全血细胞减少,累及≥2个细胞系,骨髓增生异常或增生减少;③肝功能异常及凝血障碍,纤维蛋白原(FIB)≤1.5g/L;④嗜血细胞占骨髓有核细胞的2%以上或(和)累及骨髓、肝脾、淋巴结及中枢神经系统的细胞学改变,血清铁蛋白常显著增高,是疾病活动性的一个重要指标。此外,HPS患者还应与恶性组织细胞增生症及朗格汉斯细胞增生症相鉴别[5]。本例患者既往有系统性红斑狼疮病史,在原有疾病的基础上继发了整个系统的损害,除未触及肝脾肿大外,均符合嗜血细胞综合征诊断条件,而此次发病前有明确的狼疮活动,且多次血培养表现为阴性,故考虑为狼疮急性活动时并发的HPS。本病临床症状表现严重,且可能因为治疗用药因素出现许多药物相关的副作用,造成诊断以及治疗上的困难。
HPS的预后取决于其原发病类型,据文献报道IAHS死亡率为20%~40%,非IAHS几乎100%死亡[6]。对HPS的治疗方法[4]包括有化疗,如依托泊苷(VP-16)以及系统使用糖皮质激素、环孢素A、长春新碱等免疫疗法,尚有较多文献报道[7],用大剂量丙种球蛋白的治疗。原发性HPS的根本治疗是同种异体造血肝细胞移植,而继发性HPS的原发病治疗极为重要,轻者控制原发病和加强支持治疗可康复。本例患者采用糖皮质激素、免疫制剂及抗感染联合治疗,仍反复出现血象三系减低症状,并出现长期使用糖皮质激素的副作用,对症治疗持续随访中。
[1]诸福棠. 实用儿科学 [M]. 6版. 北京:人民卫生出版社, 1997:2466-2467.
[2]Papo T, Andrc MH, Amoura Z, et al. The spectrum of reactive hemophagocytic syndrome in systemic lupus erythematosus [J].J Rheumatol, 1999, 26 (4):927-930.
[3]李文益. 儿科学新理论新技术 [M]. 北京:人民卫生出版社,2002:344-347.
[4]Kaito K, KobayashiM, Katayama T, et al. Prognostic factors of hemophagocytic syndrome in adults: analysis of 34 cases [J].Eur J Haematol, 1997, 59(4):247-253.
[5]王丛笑. 成人嗜血细胞综合征1例文献复习 [J]. 医学综述. 2008,14(12):1893-1894
[6]王学文. 嗜血细胞综合征研究进展 [J]. 国外医学输血与血液学分册, 1998, 21(6):353-356.
[7]Erduran E, Gedik Y, Sen Y, et al. Successful treatment of reactive hemophagotcytic syndrome with cyclosporine A and intravenous immunoglobulin [J]. Turk J Pediatr, 2000, 42(2):168-170.
猜你喜欢
特产研究(2022年6期)2023-01-17
临床误诊误治(2021年12期)2021-12-04
河南科学(2020年3期)2020-06-02
云南医药(2019年3期)2019-07-25
铜仁学院学报(2018年6期)2018-07-05
山东医药(2015年13期)2016-01-12
中国继续医学教育(2015年3期)2016-01-06
实用皮肤病学杂志(2015年4期)2015-12-22
医学研究杂志(2015年4期)2015-06-10
西南军医(2015年5期)2015-01-23
