
非负载型加氢精制催化剂的制备及工业应用研究进展
2010-10-19 06:42:12张景成刘晨光
化工进展 2010年4期
刘 迪,张景成,刘晨光
(中国石油大学重质油国家重点实验室,CNPC催化重点实验室,山东 青岛 266555)
进展与述评
非负载型加氢精制催化剂的制备及工业应用研究进展
刘 迪,张景成,刘晨光
(中国石油大学重质油国家重点实验室,CNPC催化重点实验室,山东 青岛 266555)
从催化剂的制备技术、催化活性等方面对新一代非负载型加氢精制催化剂进行了综述,并简要地介绍了其工业应用情况,指出了其优势和局限。工业实践证明,非负载型加氢精制催化剂活性更高,能够满足人类对清洁油品的需求。文章最后还对其制备及应用前景进行了展望。
清洁油品;非负载型;加氢精制催化剂
油品的脱硫脱氮一直是炼油工作者所面临的重大课题之一,特别是随着原油的不断开采,原油重质化、劣质化现象日益严重;同时环保法规对油品质量要求越来越苛刻,如《世界燃油规范》Ⅳ档就规定:柴油中硫含量不得超过10 µg/g,总芳烃含量不得超过15%(质量分数,下同),多环芳烃含量不得超过2%,十六烷值不得低于55等[1],所以生产出符合环保要求的超清洁油品势在必行。通过改变工艺流程、操作条件可以满足油品深度脱硫、脱氮的要求,但势必增加运营成本。因此,开发新型高效的深度加氢精制催化剂成为解决这一难题的最为经济有效的方法[2]。
目前工业上所用的加氢精制催化剂多为负载型催化剂。随着催化理论的日益完善、制备技术的进步,负载型催化剂活性也在不断提高[3],然而由于负载型催化剂中有效的活性金属负载量受到载体比表面积和孔体积的约束,难以在现有基础上大幅增加,使催化剂活性的提高受到限制。所以传统的负载型催化剂已很难同时满足深度加氢脱硫、脱氮和芳烃加氢饱和的技术要求。而非负载型催化剂不使用载体,所以其活性组分含量更高,活性位密度更大,具有很强的加氢脱硫、脱氮和芳烃饱和能力。2001年,Albemarle、ExxonMobil和Nippon Ketjen公司共同研制的NEBULA系列非负载型加氢精制催化剂成功地实现工业化[4]。运行结果表明,NEBULA系列催化剂活性为常规催化剂的2~4倍,显示出了广阔的应用前景[5]。本文就非负载型加氢精制催化剂目前的研究现状以及工业应用情况进行简要的论述。
1 非负载型加氢精制催化剂的制备
非负载型加氢精制催化剂是近几年才发展起来的一种新型的催化剂。所谓非负载型加氢精制催化剂(有些资料称之为体相催化剂或本体催化剂)是相对于传统的负载型加氢精制催化剂而言的,主要是通过增加催化剂上活性位的密度从而大大地提高其催化活性。在组成上,最大的特点是不使用载体,全部是活性组分,这样就使得单位质量或体积催化剂的催化活性达到最大,从而达到深度脱硫脱氮的目的。根据目前的研究,可将非负载型加氢精制催化剂分为两类:一类称为硫化态非负载型加氢精制催化剂;另一类为氧化态非负载型加氢精制催化剂。硫化态非负载型加氢精制催化剂指的是以含硫的过渡金属盐为原料(如四硫代钼酸铵、四硫代钨酸铵等)制备的一类催化剂,这类催化剂在开工前无需专门的硫化步骤,可直接使用;而氧化态非负载型加氢精制催化剂则是以钼酸铵、钨酸铵为原料来制备,如传统的负载型催化剂一样,开工前需预硫化。以下就这两类催化剂的制备方法及其催化性能作一简要介绍。
1.1 硫化态非负载型加氢精制催化剂
硫化态非负载型加氢精制催化剂主要的制备方法是过渡金属硫代盐的热分解法[6-8]。此法已被广泛地应用于MoS2[9-11]、WS2[12]和双组分[13-14]以及三组分催化剂[15]。过渡金属盐硫代热分解法可以细分为3种,即水热合成、器内合成、器外合成。
1.1.1 水热合成
水热合成是近几年发展起来且已被广泛应用的一种合成方法。虽然已广泛应用,但尚未有严格统一的定义。一般认为,水热合成是指在密封体系中,以水为溶剂,在温度高于室温和压力高于大气压力下,原始混合物进行非均相化学反应的一种合成方法[16]。
Yoosuk等[17]采用水热法合成出了具有高比表面积的硫化态非负载型加氢精制催化剂。大体步骤为:首先配制 (NH4)2MoS4溶液,向此溶液中加入少量的十氢化萘。再将Co(NO3)2·6H2O 或者Ni(NO3)2·6H2O溶于尽可能少的水中配成溶液,然后将两溶液加到反应器中,通氢气至初始压力2.8 MPa,密封,放到沙浴350 ℃反应2 h,即得产品。此种方法制得的催化剂具有很高的比表面积,MoS2可达到320 m2/g,NiMoS2、CoMoS2也能达到200 m2/g左右。分析显示,助剂Ni或Co的加入,降低了MoS2层的长度,增加了MoS2层的层数和曲度(图1),而MoS2层的层数和曲度通常被认为对于其活性是至关重要的[18]。加氢脱硫测试表明,助剂的加入不仅提高了催化剂的活性,还会影响含硫化合物的反应路径,即直接脱硫(DDS)和加氢脱硫(HYD)。

图1 MoS2和NiMoS2的HRTEM照片[17]
1.1.2 器外合成
在器外合成技术中,首先以钼或钨的硫代铵盐及助剂为原料,制备催化剂前体,然后将前体在还原性气体(H2S/H2,大气压力)氛围下,加热分解同时催化剂从氧化态转变为硫化态。
Huirache Acuna等[19]以四硫代钼酸铵、四硫代钨酸铵和一些有机胺为原料,首先制备出了Ni(Co)/(R4N)4MoWS8前体(R=H、CH3、C3H7),然后在15%H2S/H2的气氛中,加热分解硫化,得到Ni(Co)-Mo-W三组分硫化态非负载型催化剂,并以二苯并噻吩(DBT)为模型化合物测试了其加氢脱硫性能。表征结果显示,采用不同的有机胺模板剂所制备的催化剂的比表面积相差很大。对于Ni-Mo-W型催化剂,当R=H时的比表面积最大,R=C3H7时的比表面积最小。从表1可以看出,随着有机胺中碳链的增长,催化剂的反应速率常数及HYD/DDS选择性呈下降趋势。Zhang等[20]还利用液液、液固固液等不同的路线制备了Ni-W硫化态非负载催化剂,液液路线制备的催化剂活性最高。Genuit等[21]利用器外合成方法制备了一系列Ni(Co)-Mo硫化态非负载催化剂。作者在前体制备过程中加入了乙二醇和表面活性剂,并考察了不同的表面活性剂对催化剂比表面积和催化活性的影响。结果表明,表面活性剂的加入,能够有效地提高催化剂的比表面积和催化活性,加入Triton X114时制备的催化剂活性最高。此法制备的Ni(Co)-Mo双组分催化剂对噻吩和4,6-二甲基二苯并噻吩的脱硫活性是工业应用的负载型催化剂的6倍。同时,作者还研究了钼源对催化剂活性的影响。加氢脱硫实验显示,以 (NH4)2Mo2S12为原料制备的催化剂的脱硫活性高于以 (NH4)2MoS4为原料制备的催化剂。作者最后指出,虽然此种方法制备的硫化态非负载催化剂的活性远高于工业用负载型催化剂,但其本质活性仍然比工业用负载型催化剂低,这主要是因为本体催化剂中金属活性组分没有得到充分利用。另外,还有一些其它报道,方法基本类似,不再赘述[22-24]。此种方法制备硫化态催化剂制备过程较复杂,成本较高,难以进入工业应用。利用廉价的工业原料,无毒的硫源,简化制备步骤等是下一步重点研究的方向。

表1 Ni(Co)-Mo-W硫化态非负载型催化剂的反应速率常数,DBT转化率及HYD/DDS选择性[19]
1.1.3 器内合成
器内合成实际上是水热合成演变来的一种方法。典型制备步骤是配制一定量的四硫代钼酸铵和/或四硫代钨酸铵溶液,向混合溶液中加入模板剂(一般为有机胺等表面活性剂),再加入助剂(Co或Ni的盐溶液),即生成沉淀,将沉淀过滤,干燥,便得到催化剂前体。将催化剂前体和原料装入反应器中,进行加氢脱硫反应。这种方法最显著的特点是催化剂的制备与原料的加氢精制步骤是同时进行的。
Nava等[25]通过在DBT加氢脱硫的过程中将Ni-Mo-W三金属的烷基取代前体[(R4N)4(MoS4)3,R=H、CH3、C4H9、C5H11、C6H13]器内活化而制得非负载的介孔Ni–Mo–W硫化物催化剂。表征结果显示所得催化剂具有高比表面积、高碳含量及第Ⅳ类吸附-脱附等温线等特点,并且是介孔结构(表2)。前体中的烷基对催化剂的比表面积和催化活性的影响很大。XRD表明这些催化剂的晶态结构很弱,并发现当使用含碳的前体时催化性能会大大增强。DBT加氢脱硫结果显示,前体Co/[N(C4H9)4]2MoS4器内活化后所得的Co-Mo硫化物催化剂HDS活性最高,前体Co/[N(C6H13)4]2MoS4器内活化后所得的Co-Mo硫化物催化剂具有最高的比表面积。Nava等[26]通过PxNi/[(CH3)4N]4MoWS8制备了P-Ni-Mo-W非负载型催化剂。表3列出不同P/Mo摩尔比对Ni-Mo-W非负载催化剂加氢脱硫活性的影响。结果显示,随着磷含量的增加,催化剂的加氢脱硫性能明显降低,说明磷的加入强烈地抑制了催化剂的加氢脱硫活性。作者认为其原因是一方面磷的加入造成了孔的阻塞,大大地降低了催化剂的比表面积和孔容;另一方面,XRD研究显示,和未加磷的催化剂相比,MoS2层或WS2层的层数偏低,即Ⅱ型活性相比例较小。
过渡金属硫代盐热分解法是制备硫化态非负载型催化剂最为普遍的方法。该法制备的催化剂的比表面积可以从几平方米到几百平方米不等,催化性能相差很大,这主要取决于制备条件[27-28]。值得一提的是,过渡金属硫代酸盐中已存在呈四面体配位的M—S键,在其分解的过程中M—S键保持不变,而体相的氧化态催化剂要完全硫化是相当困难的,这是硫化态催化剂的一个很大的优势。目前,硫化态非负载型加氢精制催化剂研究还处于实验室阶段,相关研究主要集中于催化剂的制备、表征分析、反应机理以及活性等方面,要走向工业应用还有简化制备步骤、降低生产成本、解决成型强度等一系列问题需要解决。
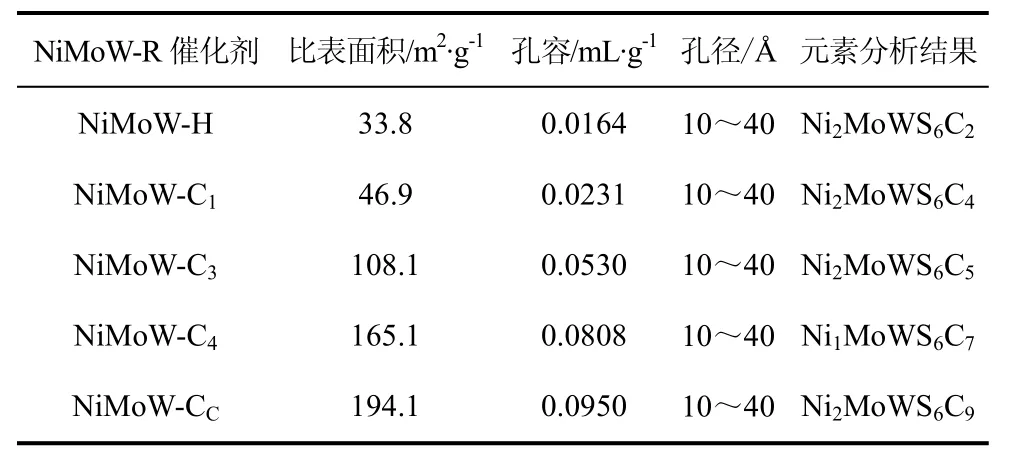
表2 NiMoW-R硫化态催化剂的比表面积、孔容、孔径分布及元素分析[25]

表3 NiMoW-P和NiMoW催化剂对于DBT加氢脱硫反应的初始反应速率常数及选择性[26]
1.2 氧化态非负载型加氢精制催化剂
根据目前的文献报道,氧化态非负载型加氢精制催化剂制备方法大体可分为两类:一是共沉淀法;二是固相反应法。与硫化态非负载型加氢精制催化剂相比,氧化态非负载型加氢精制催化剂制备工艺相对简单、成本低,已进入工业应用阶段。
1.2.1 共沉淀法
所谓共沉淀法是指所有的金属组分原料均呈溶液状态,通过调节混合溶液的pH值或改变温度等手段使反应物发生反应而形成沉淀,制得非负载型催化剂的方法。
Domokos 等[29]利用过渡金属组分盐溶液共沉淀制备出了非负载催化剂,首先配制一定量的可溶性钼酸盐(如钼酸铵)和镍盐(如硝酸镍)混合溶液,加热至80 ℃,用硝酸调节pH值到2.8,得到一澄清溶液,然后将分散于水中的二氧化硅添加到此溶液中;之后缓慢地添加氨水溶液至pH值6.8,将生成沉淀过滤、干燥,得到催化剂前体。再向前体中加入黏结剂,经过成型、焙烧等一系列步骤,就得到成品催化剂。此种方法制得的催化剂具有较大的比表面积和较高的活性,而且催化剂的机械强度完全满足工业的要求。专利[30]也介绍了通过共沉淀法来制备非负载型催化剂的方法。根据操作步骤不同,作者又将其细分为3种:沸腾分解法、直接沉淀法、控制pH值法。这3种方法大同小异,通过加入的碱液,调节溶液的pH值或者改变温度,或者改变加料顺序,将钼、钨、镍从其可溶性盐混合溶液中沉淀出来,生成Ni-Mo-W复合物,即催化剂前体。然后经过成型、焙烧等步骤,得到氧化态非负载型催化剂。经实验测试表明,此方法制得的非负载型催化剂的加氢脱硫、加氢脱氮、芳烃饱和能力活性远高于传统的负载型催化剂。中国石化石油化工科学研究院毕云飞等[31]也采用共沉淀法制备了非负载型Ni-Mo/Ni-W双组分非负载型加氢精制催化剂,不同的是在得到催化剂前体后,进行水热处理。经水热处理后的催化剂具有更高的比表面积和孔容。和参比催化剂相比,它们的活性更高。在其另一篇专利CN101306374A[32]中用了相同的方法制备Ni-Mo-W三组分非负载型催化剂。区别为在共沉淀过程中加入了有机胺、十二烷基磺酸钠等表面活性剂,实验结果表明这些活性剂的加入能提高催化剂的催化活性。中国石化抚顺石油化工研究院徐学军等[33-37]对非负载型加氢精制催化剂进行了较为深入的研究,并申请了多项发明专利,其开发的FH-FS非负载型Ni-Mo-W加氢精制催化剂已进入工业应用阶段。这种催化剂制备的思路是首先利用共沉淀法制备Ni-W复合物,让Ni-W紧密地结合在一起,然后与MoO3、黏结剂等复合。实验结果显示此种方法制备的催化剂比Ni-Mo-W三组分一起沉淀制备的催化剂的活性高。
1.2.2 固相反应法
这里固相反应法是指在催化剂制备过程中,至少一种金属组分是以固态或者部分固态的方式加入到反应体系中,并且这种金属组分要在整个反应过程中都要至少部分处于固态。同时为了获得高活性则要求处于固态的金属组分应是多孔的,其总孔容及孔径分布应与传统的加氢精制催化剂大体相同。
专利[38]介绍了一种固相合成方法。大体步骤如下:配制一定量的钼盐和钨盐混和溶液,然后将此溶液加热到90 ℃(溶液A);将一定量不溶于水的镍盐(如碱式碳酸镍)与水混合并加热到90 ℃(悬浮液B)。将悬浮液B加到溶液A中,混合后温度要仍保持在90 ℃并不停地搅拌18~20 h。完成后将悬浮液过滤、洗涤、干燥,再经过成型、焙烧等步骤,得到氧化态非负载型催化剂。此法制备的催化剂有较高的比表面积和孔容,但作者没有给出具体的活性数据。李灿等[39]在固相反应过程中加入乙二醇及表面活性剂,如十六烷基三甲基氯化铵、Triton X-100、Triton X-114或它们的组合,反应后经过过滤、干燥、焙烧、成型等步骤即得到催化剂。加氢脱硫测试表明,其活性为传统工业催化剂的5.9倍。本文作者所在课题组采用固体表面反应技术,以钼酸铵、钨酸和碱式碳酸镍为前体,制备具有微晶结构、适宜比表面积和介孔结构的Ni-Mo-W复合氧化物,并通过特有的成型技术制备出非负载型的Ni-Mo-W柴油加氢催化剂。结果表明:采用固体表面反应技术可以制备出非负载型催化剂,其中Ni-Mo-W复合氧化物的比表面积可达 113 m2/g,孔容可达0.23 cm3/g,平均孔径为7.5 nm。成型后的非负载型催化剂的颗粒平均机械强度可达120 N/cm,符合工业催化剂对强度的要求。模型化合物的加氢微反评价结果表明:所制备出的负载型Ni-Mo-W柴油加氢催化剂的HDS、HDN和HDAr活性均显著高于FH-98催化剂,显示出良好的应用前景。
催化剂性能的好坏,关键在于活性组分的选择、活性组分的结合方式等是否处于最佳状态。其中包括活性组分的选取、组成、分散方法及性能的修饰等多个过程。这一步骤主要决定催化剂的活性、选择性、催化反应的稳定性等重要指标。对于非负载型加氢精制催化剂,这些方面的认识还远远不够深入。此外,较大的比表面积和孔容、适宜的孔径是催化剂具有高活性的前提,高的比表面积与孔容能够提高活性组分的利用率;而适宜的孔径有利于反应物在催化剂中的扩散,提高反应活性;同时高的比表面积、较大的孔容,还可以提高催化剂的抗积炭能力,延长催化剂的寿命。传统的负载型催化剂都具有较大的比表面积和孔容,这主要是由其载体提供的。而对于非负载型催化剂,如何使催化剂自身具有高的比表面积,适宜的孔径、孔容就变得非常重要,也是催化剂在制备过程中的难点所在。
2 非负载型加氢精制催化剂的工业应用
非负载型加氢精制催化剂目前处于发展阶段,工业应用得还很少。国内有中国石化抚顺石油化工研究院研制的FH-FS非负载型加氢精制催化剂。FH-FS催化剂是以Ni-Mo-W 为活性组分的非负载型加氢精制催化剂,具有活性金属含量高、分散均匀、加氢脱硫脱氮活性高、稳定性好、对原料适应性强等特点,其加氢脱硫、加氢脱氮性能均明显优于常规加氢催化剂[40-42]。目前已在镇海炼化分公司燃料油加氢装置上应用[43-44],初步工艺研究结果表明,FH-FS非负载型催化剂处理原料干点高、硫含量高、密度大,二次加工柴油时,表现出优异的加氢性能,可在较缓和的条件下生产硫含量满足欧Ⅳ、欧Ⅴ标准的清洁柴油产品,可以满足进口含硫原油的二次加工柴油及其混合油加氢精制的需要。由于FH-FS催化剂进入工业应用时间很短,可以利用的数据不多,其催化活性还需要在工业应用中进一步分析验证。
NEBULA系列催化剂是最具有代表性的非负载型加氢精制催化剂。NEBULA是ExxonMobil公司的注册商标。所谓的NEBULA催化剂(new bulk activity)是由Albemarle、ExxonMobil 和Nippon Ketjen 公司共同开发的,是一项具有突破性的专利技术,并于2001年成功地实现工业化[45]。现在,NEBULA系列催化剂有NEBULA-1和NEBULA-20两种。NEBULA-20是在NEBULA-1工业应用之后,根据其实际使用情况,对NEBULA-1进一步改进而研制的第二代催化剂。和NEBULA-1相比,它的本质活性更高,能够处理更重的原料。
NEBULA催化剂于2001年首次工业化之后其装置数量迅速增加,截止2005年,NEBULA催化剂已在15套装置中得到应用,主要应用于超低硫柴油的生产、加氢裂化原料预处理、煤油及石脑油加氢精制等领域。NEBULA催化剂显示出了很高的催化活性和稳定性,它可以将劣质柴油在常规加氢精制条件下(340~350 ℃、6.0~7.0 MPa、1.0~1.5 h-1、300~500氢油比)转化为符合《世界燃油规范》Ⅳ档标准的超清洁柴油,从而满足了人们生产超低硫柴油的需求。在某些装置中,由于NEBULA催化剂的本质活性太高,为了达到最优的效果,采用了NEBULA催化剂和传统的负载型催化剂(KF760 STARS)混装的方式。在同样达到8 µg/g S的条件下,和单独使用KF760 STARS催化剂相比,NEBULA+KF760体系的稳定性和氢耗相当,但其反应温度却低了15 ℃[46]。
NEBULA系列催化剂具有很高的催化活性,但工业应用方面也存在一些不利因素。首先,由于NEBULA系列催化剂的金属活性组分含量很高,即便其活性组分全是Ⅷ和Ⅵ族的金属,其价格也要比传统的负载型催化剂高出很多,对于相同的催化剂床层,其填装费用大大增加;其次,NEBULA系列催化剂氢耗较高。NEBULA系列催化剂之所以具有较强的HDS、HDN和HAD能力,就在于其较高的加氢活性,高的加氢活性导致高的氢气消耗量,从而使得企业运营成本增加。针对这一情况,Albemarle公司提出采用NEBULA催化剂与传统的负载型催化剂混装的方法,混装后在保证活性的前提下,氢耗和负载型催化剂相当,从而达到催化剂活性与氢气消耗量的最优组合。
NEBULA系列催化剂可以使炼油者在无需改装现有设备,无需增加额外投资的情况下,即可生产出超低硫柴油,或者说在现有的装置下可以处理更加劣质的原料;该技术还具有降低能耗与CO2的排放量,提高产品质量(芳烃含量进一步降低,十六烷值进一步提高,密度进一步降低)等优点。在环保法规越来越严格,油品质量要求越来越高的背景下,NEBULA系列催化剂有着广阔的应用前景。
3 结 语
非负载型催化剂显示出很高的催化活性,和传统的负载型加氢精制催化剂相比,具有非常明显的优势,它颠覆了传统催化剂的载体与活性组分概念,是催化剂组成与活性方面的一次飞跃,代表了加氢脱硫催化剂的发展趋势。目前非负载型加氢精制催化剂的历史还很短暂,所以对其结构、活性相等基础研究还很少,负载型加氢精制催化剂的各种理论是否完全适用于非负载型加氢精制催化剂也还没有定论;另外,进一步提高非负载型加氢精制催化剂的活性、降低生产成本、简化制备工艺,是下一步努力的方向。
[1] 曾榕辉,尹恩杰.直接生产清洁柴油的加氢技术[J].炼油设计,2001,31(4):17-19.
[2] 廖健,张兵,刘伯华.国外清洁燃料生产技术[J].当代石油石化,2001,9(3):28-32.
[3] 葛晖,李学宽,秦张峰,等.油品深度加氢脱硫催化研究进展[J].化工进展,2008,27(10):1490-1497.
[4] Gochi Y,Ornelas C,Alonso-Nunez G,et al. Effect of sulfidation on Mo-W-Ni trimetallic catalysts in the HDS of DBT[J]. Catal. Today,2005,107-108:531-536.
[5] Soled S L,Miseo S,Krikak R,et al. Nickel molybodtungstate hydrotreating catalysts(law444):US,6299760[P]. 2001.
[6] Ozkan U S,Zhang L,Ni S,et al. Characterization and activity of unsupported Ni-Mo sulfide catalysts in HDN/HDS reactions[J]. Energy Fuels,1994,8(4):830-838.
[7] Inamura K K,Prins R. The role of Co in unsupported Co-Mo sulfides in the hydrodesulfurization of thiophene[J]. J. Catal.,1994,147(2):515-524.
[8] Diaz G,Luna R,Baños L,et al. X-ray diffraction study of a CoMo sulfide obtained by the impregnated thiosalt decomposition method[J]. Catal. Lett.,1990,7(5-6):377-382.
[9] Alonso G,Del Vallea M,Fuentes S. Preparation of MoS2catalysts by in situ decomposition of tetraalkylammonium thiomolybdates[J]. Catal. Today,1998,43(1-2):117-122.
[10] Alonso G,Berhault G,Aguilar A,et al. Preparation of MoS2catalysts by in situ decomposition of tetraalkylammonium thiomolybdates[J]. J. Catal.,2002,208(2):359-369.
[11] Del Valle M,Cruz-Reyes J,Fuentes S,et al. Hydrodesulfurization activity of MoS2catalysts modified by chemical exfoliation[J]. Catal. Lett.,1998,54(1-2):59-63.
[12] Breysse M,Frety R ,Lacroix M,et al. Comparison of the catalytic properties in hydrodesulfurization reaction of unsupported MoS2and WS2catalysts:Influence of surface areas[J]. React. Kinet. Catal. Lett.,1984,26(1-2):97-101.
[13] Nava H,Ornelas C,Alonso G,et al. Cobalt-molybdenum sulfide catalysts prepared by in situ activation of bimetallic(Co-Mo)alkylthiomolybdates[J]. Catal. Lett.,2003,86(4):257-265.
[14] Poisot M,Bensch W,Fuentes S,et al. High activity Ni/MoS2catalysts obtained from alkylthiometalate mixtures for the hydrodesulfurization of dibenzothiophene[J]. Catal. Lett.,2007,117(1-2):43-52.
[15] Olivas A,Galván D H,Alonso G,et al. Trimetallic NiMoW unsupported catalysts for HDS[J]. Appl. Catal. A,2009,352(1-2):10-16.
[16] Roy R. Accelerating the kinetics of low-temperature inorganic syntheses[J]. J. Solid State Chem.,1994,111(1):11-17.
[17] Yoosuk B,Kim J H,Song C,et al. Highly active MoS2,CoMoS2and NiMoS2unsupported catalysts prepared by hydrothermal synthesis for hydrodesulfurization of 4,6-dimethyldibenzothiophene[J]. Catal. Today,2008,130(1):14-23.
[18] Iwata Y,Sato K,Yoneda T,et al. Catalytic functionality of unsupported molybdenum sulfide catalysts prepared with different methods[J]. Catal. Today,1998,45(1-4):353-359.
[19] Huirache Acuna R,Albiter M A,Ornelas C,et al. Ni(Co)-Mo-W sulphide unsupported HDS catalysts by ex situ decomposition of alkylthiomolybdotungstates[J]. Appl. Catal. A,2006,308:134-142.
[20] Zhang L,Afanasiev P,Li D,et al. Solution synthesis of the unsupported Ni-W sulfide hydrotreating catalysts[J]. Catal. Today,2008,130(1):24-31.
[21] Genuit D,Afanasiev P,Vrinat M. Solution syntheses of unsupported Co(Ni)-Mo-S hydrotreating catalysts[J]. J. Catal.,2005,235(2):302-317.
[22] Álvarez L,Berhault G,Alonso-Nuñez G. Unsupported NiMo sulfide catalysts obtained from nickel/ammonium and nickel/ tetraalkylammonium thiomolybdates:synthesis and application in the hydrodesulfurization of dibenzothiophene[J]. Catal. Lett.,2008,125(1-2):35-45.
[23] Olivas A,Galván D H,Alonso G. Trimetallic NiMoW unsupported catalysts for HDS[J]. Appl. Catal. A,2008,352(1-2):10-16.
[24] Olivas A,Zepeda T A,Villalpando I,et al. Performance of unsupported Ni(Co,Fe)/MoS2catalysts in hydrotreating reactions[J]. Catal. Commun.,2008,9(6):1317-1328.
[25] Nava H,Pedraza F,Alonsoa G. Nickel-Molybdenum-Tungsten Sulphide catalysts prepared by in situ activation of trimetallic(Ni-Mo-W)alkylthiomolybdotungstates[J]. Catal. Lett.,2005,99(1-2):65-71.
[26] Nava H,Espino J,Alonso Nunez G. Effect of phosphorus addition on unsupported Ni-Mo-W sulfide catalysts prepared by the in situ activation of nickel/tetramethylammonium thiomolybdotungstate[J]. Appl. Catal. A,2006,302(2):177-184.
[27] Alonso G,Aguirre G,Fuentes S,et al. Synthesis and characterization of tetraalkylammonium thiomolybdates and thiotungstates in aqueous solution[J]. Inorg. Chim. Acta.,1998,274(1):108-110.
[28] Ramanathan K,Weller S. Characterization of tungsten sulfide catalysts[J]. J. Catal.,1985(1),95:249-259.
[29] Domokos L,Jongkind H,Van veen J A R. Catalyst composition preparation and use:WO,2004/073859[P]. 2004.
[30] Demmin R A,Riley K L. Hydrconversion process using bulk group VIII/group VIB catalysts:WO,2000/042119[P]. 2000.
[31] 毕云飞,曾双亲,聂红,等. 一种非负载型加氢催化剂的制备方法:中国,101468309[P],2007.
[32] 张乐,龙湘云,李大东,等. 一种加氢催化剂组合物、制备及其应用:中国,101306374[P]. 2008.
[33] 徐学军,王继锋,刘东香,等. 一种加氢催化剂的制备方法:中国,101172261[P]. 2008.
[34] 徐学军,冯小萍,王继锋,等. 催化剂组合物的制备方法:中国,1951558[P]. 2007.
[35] 徐学军,刘东香,王继锋,等. 一种催化剂组合物的制备方法:中国,1951559[P]. 2007.
[36] 徐学军,刘东香,王继锋,等. 加氢催化剂组合物的制备方法:中国,1952054[P]. 2007.
[37] 徐学军,王海涛,王继锋,等.一种加氢催化剂组合物的制备方法:中国,1952057[P]. 2007.
[38] Eijsbouts S,Oogjen B G,Homan Free H W,et al. A mixed metal catalyst composition,its preparation and use:WO,200041810[P]. 2000.
[39] 李灿,蒋宗轩,王璐.用于柴油加氢脱硫的多金属本体催化剂及制法和应用:中国,101153228[P]. 2008.
[40] 李扬,王震,徐学军,等. 一种生产超低硫柴油方法:中国,101280216[P]. 2007.
[41] 刘涛,田洪良,曾榕辉,等.一种加氢处理方法:中国,101089133[P]. 2007.
[42] 刘涛,赵玉琢,方向晨,等.一种柴油馏分加氢改质方法:中国,101089131[P]. 2007.
[43] 王震,徐学军,史开洪,等.汽、煤油混合加氢技术开发及工业应用[J].炼油工程与技术,2008,38(4):5-8.
[44] 黄叔儒. FH-40C及FH-FS催化剂在镇海炼化分公司燃料油加氢装置上的应用[J].石化技术与应用,2009,27(1):36-40.
[45] Song C. An overview of new approaches to deep desulfurization for ultra-clean gasoline,diesel fuel and jet fuel[J]. Catal. Today,2003,86(1-4):211-263.
[46] Eijsbouts S,Mayo S W,Fujita K. Unsupported transition metal sulfide catalysts:From fundamentals to industrial application[J]. Appl. Catal. A,2007,322:58-66.
Progress in preparation and industrial application of unsupported hydrotreating catalysts
LIU Di,ZHANG Jingcheng,LIU Chenguang
(State Key Laboratory of Heavy Oil Processing,Key Laboratory of Catalysis,CNPC,China University of Petroleum,Qingdao 266555,Shandong,China)
This review summarizes the preparation,catalytic activity and industrial application of new unsupported hydrotreating catalysts. The advantages and limitations of unsupported hydrotreating catalysts are discussed. Industrial application has proved that unsupported hydrotreating catalysts show higher catalytic activity than supported ones and can meet the requirements for clean fuel. Finally,prospects for the synthesis and application of unsupported hydrotreating catalysts are discussed.
ultra-clean oil;unsupported;hydrotreating catalysts
O 643.38;O 612.6
A
1000–6613(2010)04–0643–07
2009-09-10;修改稿日期:2009-11-13。
国家973计划资助项目(2010CB226905)。
刘迪(1979-),博士研究生。联系人:刘晨光,教授。电话0532-86981716;E-mail cgliu@upc.edu.cn。
猜你喜欢
天津医科大学学报(2021年1期)2021-12-05 11:11:05
中国特种设备安全(2019年3期)2019-04-22 05:05:38
中国特种设备安全(2018年10期)2018-12-18 02:17:16
黑龙江科学(2017年21期)2017-12-14 08:39:34
山西化工(2016年5期)2016-12-17 08:13:15
山东工业技术(2016年15期)2016-12-01 05:30:43
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
环境科技(2015年5期)2015-11-08 12:08:58
河南科技(2014年15期)2014-02-27 14:12:38
茶叶通讯(2014年2期)2014-02-27 07:55:40
