
毛细管电泳电致化学发光法检测利多卡因
2010-10-10 00:30杨艳梅刘征远
唐山学院学报 2010年6期
杨艳梅,刘征远
(1.承德实验中学,河北承德 067101;2.唐山师范学院化学系,河北唐山 063000)
毛细管电泳电致化学发光法检测利多卡因
杨艳梅1,刘征远2
(1.承德实验中学,河北承德 067101;2.唐山师范学院化学系,河北唐山 063000)
分析了各种分离条件和检测条件对发光强度的影响;并在实验优化的条件下,对浓度在1.0 ×10-6~1.0×10-4mol·L-1范围内的利多卡因进行检测,发现发光强度与其浓度呈良好的线性关系,线性回归方程为ΔI=11.74c+34.696(c:μmol·L-1),线性相关系数R2=0.991 7。方法的检出限为1.1×10-7mol·L-1,对1.0×10-5mol·L-1的利多卡因溶液进行10次测量,相对标准偏差为1.12%。
毛细管电泳;电化学发光;联吡啶钌;利多卡因
利多卡因(结构式如图1),一种酰胺类中效局麻药,具有起效快、弥散广、穿透性强、安全范围较大、无扩张血管作用及对组织无刺激等特点,也是抗心律失常的常用药物之一;但是当血药浓度超过5μg·mL-1时出现中毒症状,超过7μg·m L-1时出现惊厥[1],因此其血药浓度监测具有重要的临床意义。以往多采用酶免疫分析法[2]、气相和高效液相色谱法[3]。但成本高,操作繁琐,干扰多,色谱柱易受污染等。
本文基于利多卡因对联吡啶钌的电化学发光具有增敏作用,通过优化实验条件探讨了一种高效、快速、灵敏、简单的测定利多卡因的方法——毛细管电泳电致化学发光法(CE2ECL)。
1 原理
Ru(bpy)32+的发光原理:一般认为,Ru(bpy)32+由电化学氧化或还原分别产生 Ru(bpy)33+或 Ru(bpy)3+,二者再
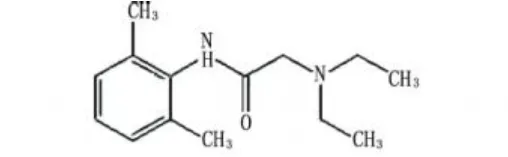
图1 利多卡因结构式

由图2可知:除刚运行时受仪器本身背景噪音影响,电流值、发光强度均较大,但在循环几圈后,循环伏安图逐渐重合,发光强度也趋于稳定;这说明 Ru(bpy)32+在此电化学条件下可循环再生,发光效率高,且化学性能稳定。
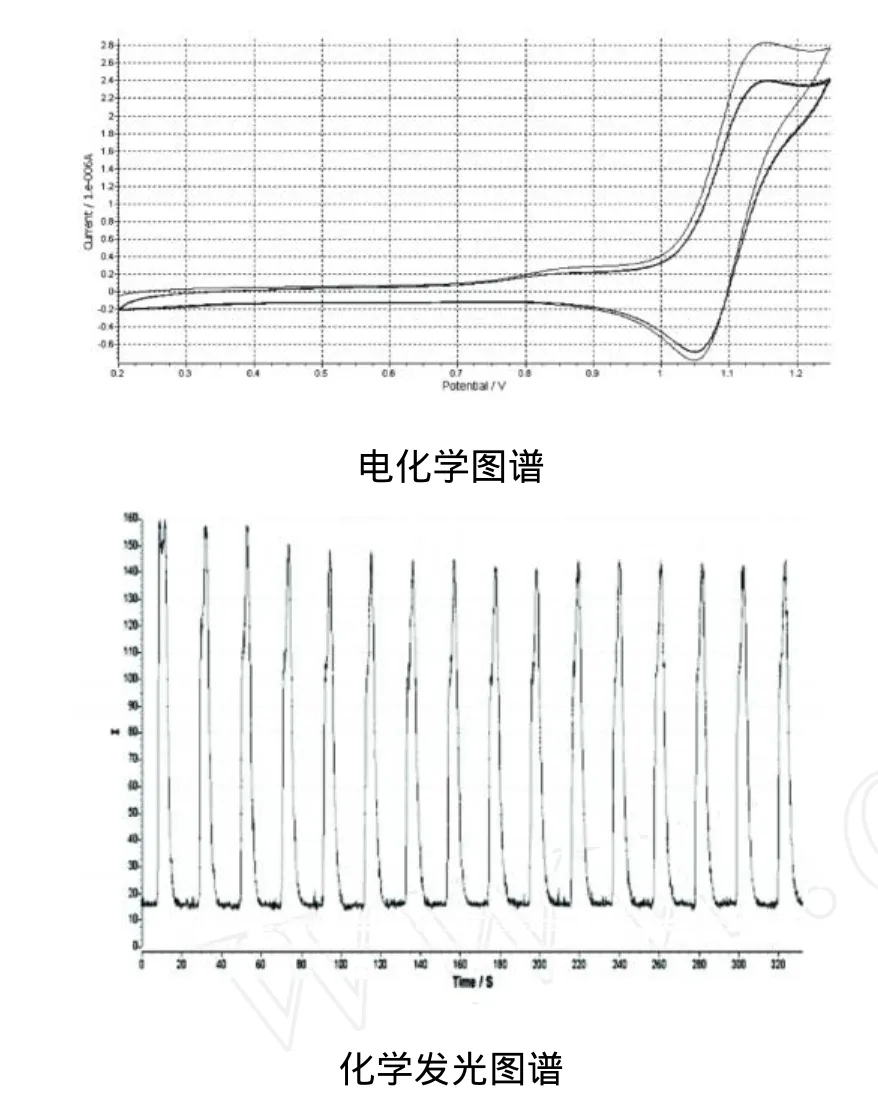
图2 Ru(bpy)32+的循环伏安图
Ru(bpy)32+本身在电极电位为1.00 V~1.25 V范围内能够进行电化学发光反应,利多卡因的加入能够促进Ru(bpy)32+3的生成,使循环伏安的氧化电流显著增大(图3);在恒电位条件下,表现为其发光强度增大,并在化学发光图谱上以峰的形式出现(图4)。
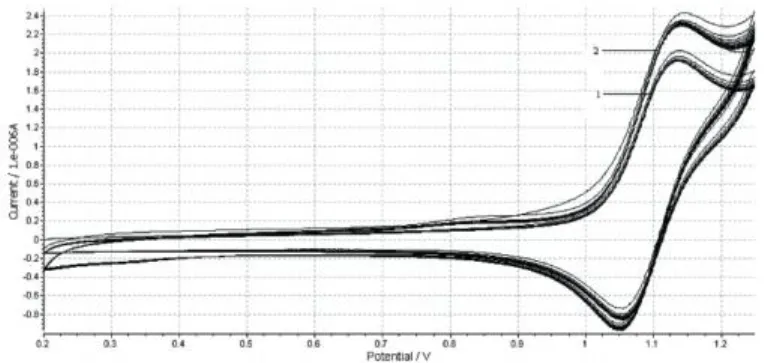
图3 循环伏安图
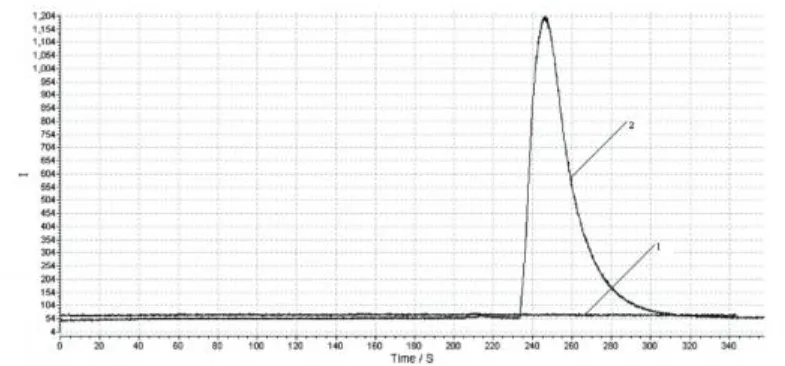
图4 化学发光图
2 实验部分
2.1 仪器与试剂
仪器:2A型多参数化学分析测试系统,未涂层石英毛细管,MOTIC体式显微镜,电子分析天平,KQ2250E型超声波清洗器,SZ293自动双重纯水蒸馏器,p HS22C型酸度计。
试剂:联吡啶钌,利多卡因,甲氧氯普胺,盐酸,磷酸,磷酸二氢钠,磷酸氢二钠,氢氧化钠,氯化钾,A lpha A lum Ina Pow der。
2.2 实验方法
2.2.1 仪器的安装
(1)毛细管的处理。毛细管在第一次使用前要按以下方法处理:依次用0.1 mol·L-1盐酸溶液浸泡30 min,二次蒸馏水(二次水)冲洗30 min;0.1 mol·L-1氢氧化钠溶液浸泡1 h,二次水冲洗30 min;磷酸盐缓冲液(PBS)(p H=8.00, 10.0 mmol·L-1)平衡4~5 h。以后每次使用前依次用二次水、氢氧化钠溶液(0.1 mol·L-1)、二次水、PBS(p H=8.00, 10.0 mmol·L-1)冲洗毛细管。所有溶液(包括上述处理溶液及待测定样品溶液)进入毛细管前均须用0.22μm醋酸纤维素膜过滤,以防止毛细管堵塞。
(2)电极的处理。工作电极处理方法:使用前将工作电极配合抛光粉(0.3μm A l2O3粉末)在抛光布上抛光,至达到镜面效果(在高倍显微镜下观察表面光滑无任何痕迹)后,用二次水进行超声清洗。
(3)检测池的安装。检测池、工作电极、辅助电极及毛细管,装入检测池;在高倍显微镜下调节使毛细管口正对工作电极,中轴线重合,并且两者距离为75μm[5,6]。将检测池装入M PI2A型多功能化学发光检测器的暗盒内(注意:在仪器运行时切勿打开暗盒,以免损坏光电倍增管!)。
(4)待测样品的装入。在进样托架上依次放入装有PBS,二次水,样品溶液的离心管,让毛细管和毛细管电泳阳极电极(铜丝电极)浸入液体中。
(5)酸度计的校准。使用p H=4.003和p H=6.864的两种标准缓冲溶液对酸度计进行校准。
2.2.2 溶液的配制
(1)联吡啶钌溶液。用电子天平准确称取0.074 9 g,用二次水配制浓度为10.0 mmol·L-1的溶液,再用磷酸盐缓冲液(p H=8.00,100.0 mmol·L-1)稀释到5.0 mmol·L-1。
(2)磷酸盐缓冲液(PBS)。用二次水配制(1)浓度为10.0 mmol·L-1,p H为6.00~8.50(每隔0.50 p H单位取一点)的一系列磷酸盐缓冲液;(2)p H=7.00,浓度为20.0 mmol·L-1~80.0 mmol·L-1的一系列磷酸盐缓冲液。上述所配磷酸盐缓冲液的p H均需要用酸度计进行校准。
(3)利多卡因储备液。用电子天平准确称取0.005 9 g,用二次水溶解,并定容于250 mL容量瓶中,得到1.00× 10-4mol·L-1的利多卡因储备液。
2.2.3 分离条件的优化实验
固定检测条件,利用上述溶液依次测量确定最佳进样电位、采样时间、进样量、分离电压,运行缓冲溶液的适宜p H和最佳浓度。
2.2.4 检测条件的优化实验
固定分离条件,利用上述溶液依次对工作电极初始电位、检测池中Ru(bpy)32+的浓度及检测池中磷酸盐缓冲液的p H和浓度进行优化。
2.2.5 工作曲线的绘制
配制浓度为1.00×10-7~1.00×10-4mol·L-1范围内的一系列利多卡因的标准溶液,在实验确定的最佳CE2ECL条件下,测定利多卡因增强的 Ru(bpy)32+发光强度(ΔI=I -I0,ΔI为增强的发光强度,I为加入利多卡因后的峰值光强,I0为背景光强(基线对应的光强),即 Ru(bpy)32+本身产生的光信号大小。),以ΔI对利多卡因的浓度c进行线性回归,绘制工作曲线。
2.2.6 利多卡因与甲氧氯普胺的分离实验
在利多卡因的CE2ECL最佳条件下,对两者混合液进行分离并检测各自发光信号。
3 实验结果与讨论
3.1 分离条件的优化
3.1.1 进样电压和时间
本实验采用电驱动进样,在允许进样量范围内,进样电压和时间直接影响进样量,从而影响发光强度δI和柱效。实验发现,ECL光强随着进样电压和进样时间的增加而增加(如图5和图6),而柱效则随着电压和时间的增加而降低,且峰形拖尾现象越来越严重。综合考虑发光强度ΔI,柱效,峰形等因素,选择进样条件为:10 kV/10 s。
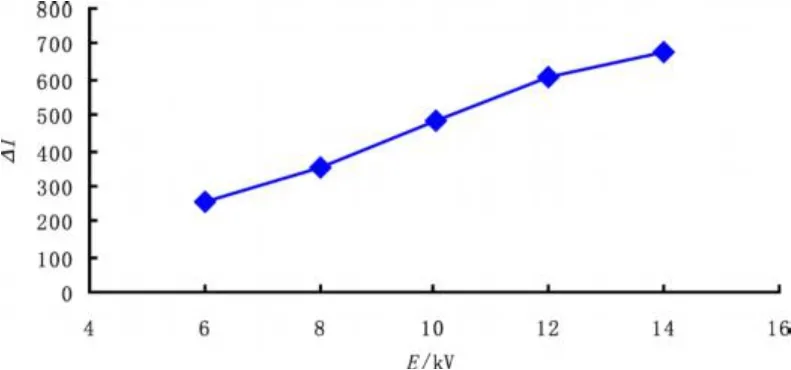
图5 进样电压对ΔI的影响
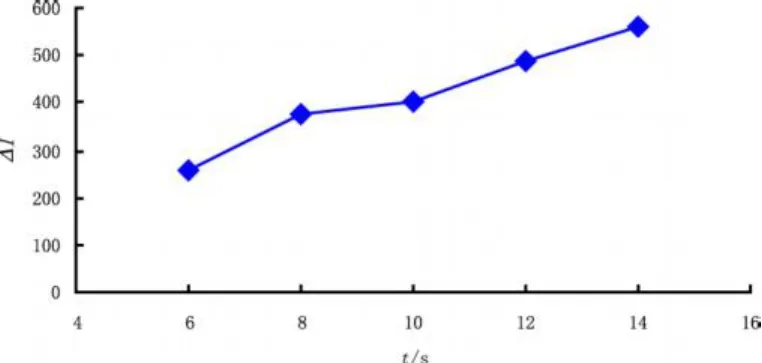
图6 进样时间对ΔI的影响
3.1.2 分离电压
实验考察了分离电压为10~18 kV时,对ΔI和出峰时间的影响。结果表明:ΔI随分离电压无明显变化,被测组分的迁移时间随分离电压的升高而缩短,但峰形逐渐变差,且拖尾现象严重,因为分离电压过高,产生的焦耳热增多。考虑迁移时间和峰形等因素,选择16 kV为最佳分离电压。
3.1.3 运行缓冲液的p H
运行缓冲液的p H直接影响毛细管表面的zeta电势,从而影响电渗流(EOF)的方向和速率,同时运行缓冲液的酸度也决定样品分子的荷电状况,从而改变分析物的迁移时间,进而对发光强度ΔI产生很大的影响[7]。当运行缓冲液的p H在6.00~8.50范围内变化时,ΔI先随着p H的增加而增大,当p H=7.00时ΔI达到最大值,而后随着p H的增大ΔI反而减小(图7)。实验选择运行缓冲液的p H值为7.00。

图7 运行缓冲液的p H对发光强度ΔI影响
3.1.4 运行缓冲液的浓度
固定运行缓冲液的p H=7.00,在10.0~50.0 mmol· L-1范围内(每隔10.0 mmol·L-1取一个点)调节缓冲液的浓度,优化运行缓冲液的浓度。实验结果表明:在较小浓度范围内,发光强度ΔI随着浓度的增大而增大,当浓度为30.0 mmol·L-1时,ΔI值最大,且峰型较好,而后ΔI反而减小(图8);另外,运行缓冲液浓度越大,出峰时间随浓度的增大而延长。综合考虑对出峰时间与发光强度的影响,选择运行缓冲液的最佳浓度为30.0 mmol·L-1。

表1 运行缓冲液浓度对出峰时间的影响
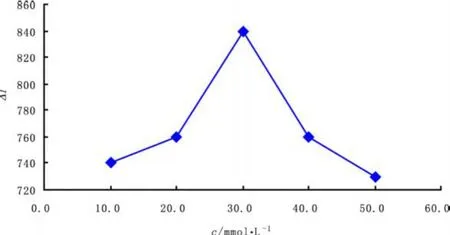
图8 运行缓冲液浓度对ΔI的影响
3.2 检测条件的优化
3.2.1 工作电极电位的优化
当电位低于1.0 V时,因Ru(bpy)32+没有被氧化,观察不到发光信号;在1.00~1.10 V间时发光信号较弱,故本实验在1.10~1.24 V范围内研究了ΔI和工作电极电位的关系(图9)。在电位较低时,ΔI随电极电位的增大而增大,在1.20 V达到最大值,而后随电位的增加反而减小,这可能是因为溶液中的水被氧化对发光强度ΔI产生负面影响造成的[8]。
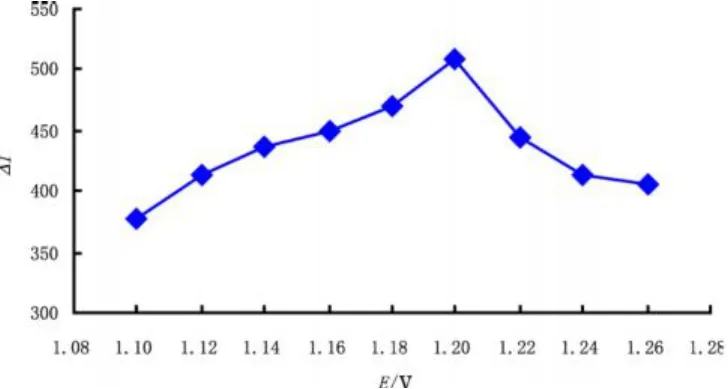
图9 工作电极电位对ΔI的影响
3.2.2 检测池中Ru(bpy)32+的浓度
由Ru(bpy)32+的化学发光反应方程式(1~4)可知:一般而言,增大检测池中Ru(bpy)32+的浓度,生成氧化态Ru (bpy)33+的量显然会增大,相应体系的发光强度也会增强;但增大浓度有时会生成沉淀,且可能增加干扰(背景噪声),且Ru(bpy)32+是比较昂贵的试剂,采用较高的浓度会增加测定成本,又因为 Ru(bpy)32+浓度为5.0 mmol·L-1时有较大的信噪比[9],所以本论文采用浓度为5.0 mmol·L-1的Ru(bpy)32+溶液为发光底液。
3.2.3 检测池中PBS浓度
本实验研究了p H为8.00的 PBS浓度在20.0~70.0 mmol·L-1(每10.0 mmol·L-1取一个点)范围内变化时对ΔI的影响。结果表明:PBS浓度在20.0~50.0 mmol·L-1范围内,ΔI随浓度的增大而增大,当其浓度为50.0 mmol·L-1时,ΔI值最大,且峰型较好;而后,发光强度ΔI反而减小(图10)。故选择检测池内PBS的最佳浓度为50.0 mmol·L-1。
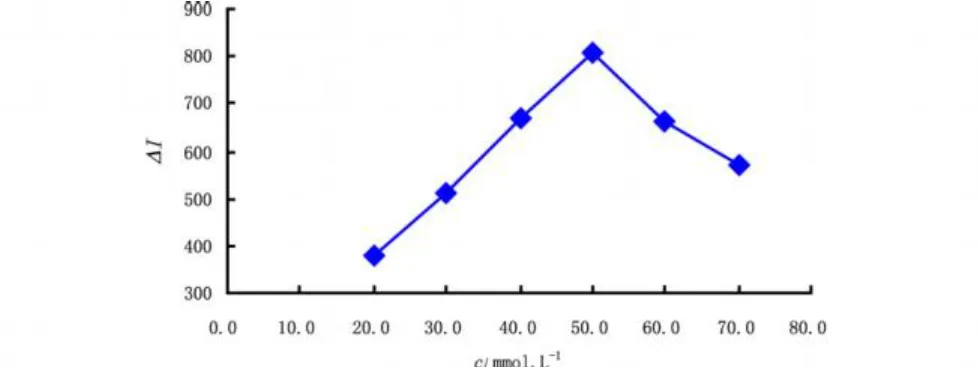
图10 检测池中Ru(bpy)32+浓度对发光强度ΔI的影响
3.2.4 检测池中PBS的p H
实验选用50.0 mmol·L-1磷酸盐缓冲液。Ru(bpy)32+和胺类物质的电化学发光反应很大程度上依赖于p H。在p H 6.00~9.00(每0.5个p H单位取一个点)范围内考察了检测池中PBS的p H对ΔI的影响(如图11)。在p H较小时,由于胺类物质质子化,ΔI小;随着p H的增大,ΔI增大,当p H为8.00时ΔI达到最大;此后,随着p H的增大,ΔI减小。故实验选用PBS的p H为8.00。

图11 检测池中PBS的p H对ΔI的影响
3.3 工作曲线、检出限和精密度的测定
3.3.1 工作曲线的绘制
按照上述实验方法,在实验确定的最佳CE2ECL条件下,测定了一系列的利多卡因标准溶液的发光强度ΔI,并以发光强度ΔI对利多卡因的浓度c进行线性回归,绘制工作曲线。线性范围为1.00×10-6~1.00×10-4mol·L-1,回归方程为ΔI=11.74c+34.696(c:μmol·L-1),相关系数 R2=0.991 7。
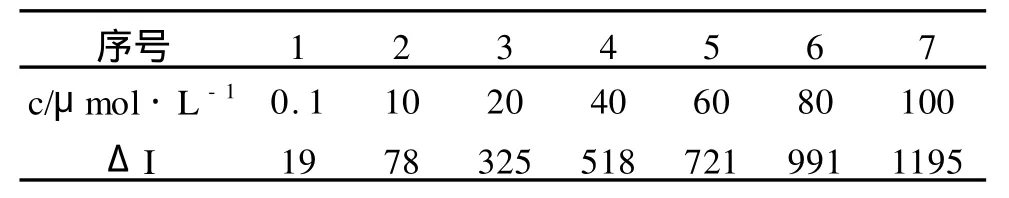
表2 标准曲线测定数据

图12 CE2ECL法测定利多卡因的标准曲线
3.3.2 精密度和检出限的测定
按照绘制工作曲线的实验方法,对1.00×10-5mol· L-1的利多卡因溶液的发光强度ΔI进行了10次测量,测得其相对标准偏差为1.12%。
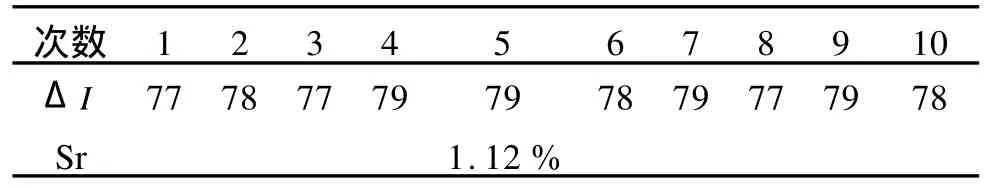
表3 1.00×10-5 mol·L-1的利多卡因溶液的ΔI
以水作为为空白试剂,按照绘制工作曲线的实验方法,进样15次,ΔI的标准偏差sb为0.45,按照IUPAC推荐的检出限的计算方法D=3sb/S,计算得在CE2ECL最佳条件下检出限为1.10×10-7mol·L-1(S为线性回归方程的斜率)。
3.4 利多卡因与甲氧氯普胺的分离与检测
甲氧氯普胺(Metoclop ramide)是一种止吐及催吐药,与利多卡因的结构相似,但两者的药理却相差很大;因此我们有必要对两者进行分离并检测。本实验在利多卡因的最佳CE2ECL条件下对利多卡因与甲氧氯普胺的浓度比为1∶100的混合液进行了分离测定,并实验了分离电压为12.0~18.0 kV时对两者分离度及发光信号的影响(表4)。
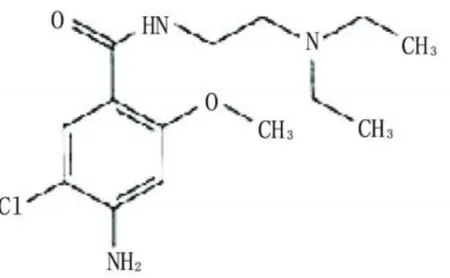
图13 甲氧氯普胺结构
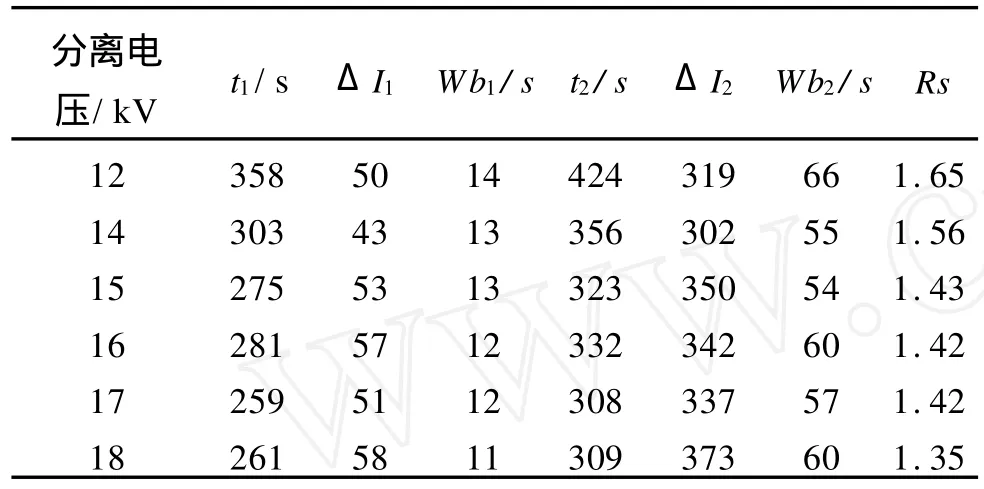
表4 分离电压对甲氧氯普胺(ΔI1)与利多卡因发光信号(ΔI2)及分离度的影响
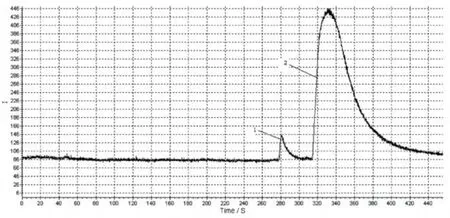
图14 利多卡因与甲氧氯普胺的CE2ECL图
由表6可知,分离电压对混合组分的迁移时间、发光强度、峰宽及分离度都有很大影响。随分离电压的升高,被测组分的发光信号逐渐变大,迁移时间缩短,但峰形有变宽趋势,且拖尾现象严重,因此分离度变差。这是由于毛细管的表面积与体积之比不足够大,散热就不够良好,而管壁与管中心的温差使溶液粘度改变,使正常的电渗流流型受到扰动,向流体动力学流型转变,使峰展宽,因此分离度降低[8],图14是利多卡因与甲氧氯普胺的CE2ECL图。由CE2ECL图可知,二者在测定条件下可以完全分离,因此CE2ECL具有很好的分离能力。
4 结论
本文建立了毛细管电泳电化学发光法测定利多卡因的方法,得结论如下:
(1)最佳分离条件为:进样条件为10 kV/10 s,分离电压取16 kV,运行缓冲液采用p H=8.00、30.0 mmol·L-1的磷酸盐缓冲液。
(2)最佳检测条件为:工作电极电位为1.20 V,检测池内Ru(bpy)32+浓度为5.0 mmol·L-1,检测池内磷酸盐缓冲液的p H值等于8.00,浓度为50.0 mmol·L-1。
(3)在CE2ECL最佳实验条件下,利多卡因可在较短时间内被检测,并且其发光强度ΔI与浓度呈线性关系,线性范围为1.0×10-6~1.0×10-4mol·L-1,回归方程为ΔI=11.74c +34.696(c:μmol·L-1),相关系数 R2=0.991 7,检出限为1.10×10-7mol·L-1,浓度为1.00×10-5mol·L-1的利多卡因用实验建立的方法测得其相对标准偏差为1.12%。
(4)CE2ECL方法可以有效分离利多卡因及其结构类似物甲氧氯普胺的混合液(Rs>1),说明该方法的分离能力强。
[1] 庄心良,曾因明,陈伯鉴.现代麻醉学[M].第三版.北京:人民卫生出版社,2003:951-960.
[2] Pape BE,W hiting R,Parker KM,et al.Enzyme Im-munoassay and gas2liquid chromatography compared for determination of lidocaine in serum[J].Clin Chem, 1978(24):2020-2022.
[3] Swezey CB,Ponzo JL.Anticholinergic activity in the serum of patients receiving maintenance disopyramide therapy[J].Clin Biochem,1984(17):325-329.
[4] Guo W W,Yuan J P,L IB L,Du Y,Ying EB,Wang E K.[Ru(bpy)2dppz]2+ Electrochemiluminescence Switch and Its Applications for DNA Interaction Study and Label2free A TP Aptasensor[J].Analyst,2008 (133):1209-1213.
[5] Jianguo Li,Fengjuan Zhao,Huangxian Ju.Simultaneous electrochemiluminescence determination of sulpiride and tiapride by capillary electrophoresis with cyclodex-trin additives[J].J Chromatogr B,2006(835):84-89.
[6] Cao W D,Liu J F,Yang X R et al.Electrochemical Detection for Capillary and Microchip Electrophoresis [J].Electrophoresis,2002,23(21):3683.
[7] 龚燕,孙秀兰,邵景东.毛细管电泳-电化学检测法测定鸡蛋中残留四环素类抗生素[J].食品科学,2007,28 (10):470-472.
[8] 方慧群,于俊生,史坚.仪器分析[M].第一版.北京:科学出版社,2008:194.
[9] 李海娟,韩双,胡连哲,等.联吡啶钌电化学发光研究进展[J].分析化学,2009,37(11):1557.
(责任编校:赵树文)
Detecting Attapugite with Electrochemical Luminescence by Capillary Electrophoresis
YANG Yan-mei1,LIU Zheng-yuan2
(1.Chengde Experimental Middle School,Chengde 067101,China;2.Chemical Department Tangshan Teachers College,Tangshan 063000,China)
The paper analyses the influence of various separation and detection conditions on luminous intensity and,when lidocaine is detected in the optimized experiment condition within the density between 1.0×10-6~1.0×10-4mol·L-1,it is discovered that luminous intensity are in good linear relationship with its density and equation of linear regression is△=11.74c+34.696 (c:μmol·L-1)and the linear related coefficient R2=0.9917.The detection limit of the method is 1.1×10-7mol·L-1and through 10 detections of lidocaine of 1.0×10-5mol·L-1the relative standard deviation is 1.12%.
capillary electrophoresis;electrochemical luminescence;attapugite;lidocaine
R96
A
1672-349X(2010)06-0067-05
2010-10-19
杨艳梅(1976-),女,中教一级,主要从事化学研究。
猜你喜欢
石家庄学院学报(2021年6期)2021-11-26
云南医药(2021年3期)2021-07-21
云南化工(2020年11期)2021-01-14
中西医结合心血管病电子杂志(2017年27期)2018-02-07
杂草学报(2015年2期)2016-01-04
中外医疗(2015年5期)2016-01-04
中药与临床(2015年5期)2015-12-17
右江医学(2015年1期)2015-03-20
中国当代医药(2015年30期)2015-03-01
食品工业科技(2014年5期)2014-03-11
