
一种采用知识打分函数的分子对接方法
2010-09-28 08:31王希诚,赵晓宇,康玲,李洪林
大连理工大学学报 2010年2期
王 希 诚, 赵 晓 宇, 康 玲, 李 洪 林
(1.大连理工大学工业装备结构分析国家重点实验室,辽宁大连 116024;2.大连理工大学工程力学系,辽宁大连 116024;3.大连理工大学计算机科学与技术学院,辽宁 大连 116024;4.中国科学院上海药物研究所,上海 201203)
0 引 言
随着计算机技术的高速发展及其在各个领域的广泛应用,计算机辅助药物设计已经成为创新药物研究的一种新方法和技术.分子对接作为基于受体药物设计的重要方法之一,已经成为可靠、相对廉价的用于先导化合物发现的一种重要手段.分子对接包括3个相互关联的部分:结合位点的识别、有效的构象优化方法及打分函数.20世纪80年代,Kuntz等[1]发展了模拟小分子与生物大分子结合三维结构及其强度的计算方法——分子对接(molecular docking)方法,并开发了第一个分子对接程序DOCK.此后,为得到精确的结合构象和正确地预测活性,各种构象优化方法及打分函数应运而生.
本文通过Boltzmann规则将原子间距离的概率分布转化为与距离有关的蛋白质-配体原子对间作用能的知识打分函数,将其与基于信息熵的多种群自适应遗传算法相结合,形成有效的分子对接程序,用于计算配体与蛋白质的结合能;并与DOCK6.1对接结果相比较,以证明其有效性.
1 基于知识打分的分子对接模型
目前,可以用于分子对接及虚拟筛选的结合自由能评价方法,大致上可以分为基于力场、基于经验及基于知识的3类打分函数.基于力场的打分函数多采用AMBER和CHARMM力场的非键相互作用部分,将蛋白质受体-配体的结合自由能近似为范德华力与静电力相互作用的加和,DOCK4[2](最新版本为 DOCK6.1)、GAsDock[3]等对接程序均采用力场打分函数作为分子对接的评价标准.经验打分函数认为结合自由能可以通过多项不同作用的加和来解释,权系数可以通过已知结合能的蛋白质-配体的训练集获得.知识打分函数通过已知的受体-配体结构,利用Boltzmann规则[4]将原子间距离的概率分布转化为与距离有关的受体-配体原子对间的作用能,并将结合过程中具有复杂相关性而又很难明确建模的结合效应隐含进去.本文采用类似经典打分PMF[5](potentials of mean force)的构造方法,从包含2422个复合物的训练集中确定了17种蛋白质受体原子类型(详见表 1)及 25种配体原子类型(详见表 2),通过Boltzmann规则得到了不同类型原子对在各个距离上的作用能,并且通过体积修正项将结合过程中的疏水作用及熵变隐含进去,其表达式如下:

表1 蛋白质原子类型Tab.1 Protein atom type

表2 配体原子类型Tab.2 Ligand atom type
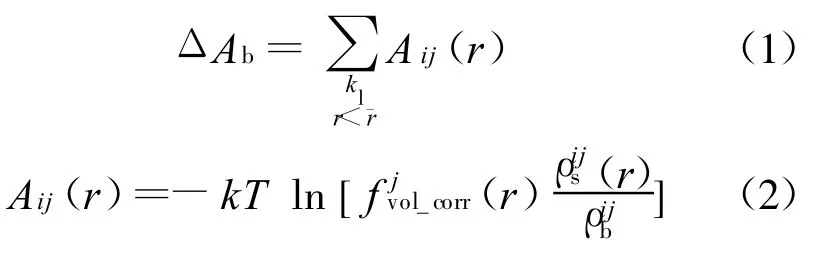
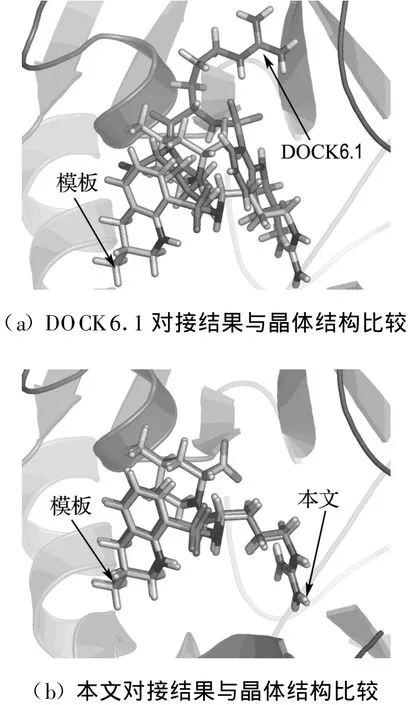
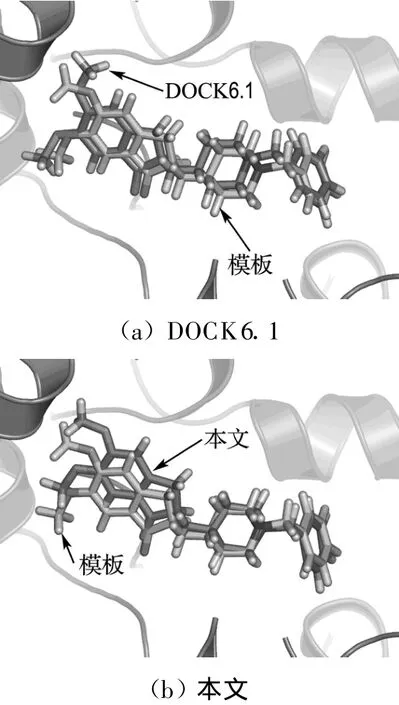
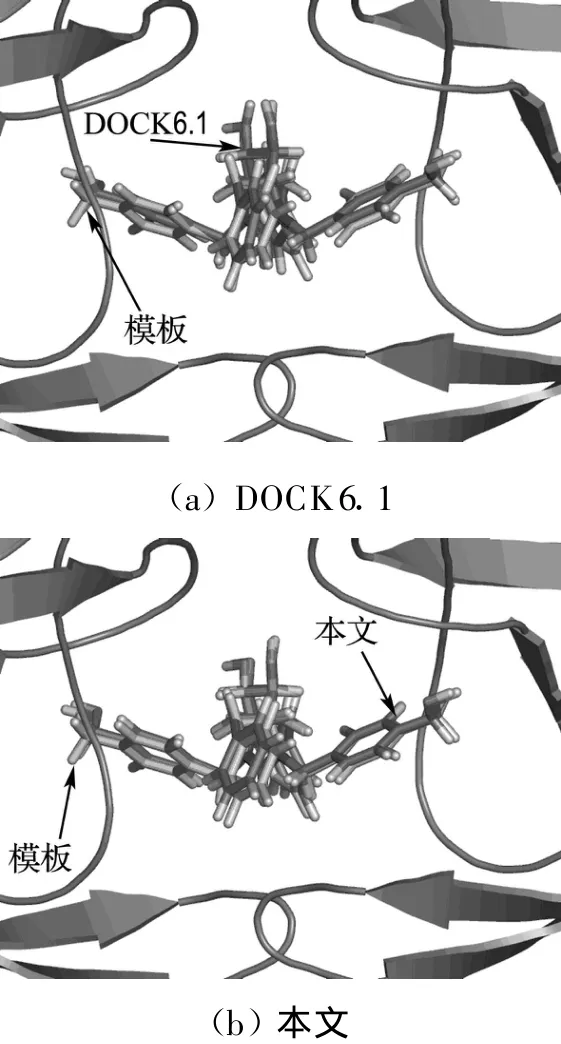
式中:Aij(r)为i类型受体原子与j类型配体原子在距离r上的能量值;kl为复合物训练集中所有距离r 本文采用只考虑小分子柔性的半柔性对接优化模型,包括小分子平动、转动及旋转键在内的一系列变化.优化对接模型为 式中:x=(TxTyTzRxRyRzTb1Tb2… Tbn)T,其中 Tx、Ty、Tz、Rx、Ry 、Rz 是配体分子的几何中心及旋转度,对应于配体分子的取向,Tb1,Tb2,…,Tbn是配体分子的可旋转键,描述配体分子的构象信息,n为可旋转键数目.目标函数 f(x)选取上述知识型打分函数. 本文在采用带有空间收缩的多种群遗传算法[6]的基础上同时加入了自适应策略,将其与知识打分函数结合用于寻找分子对接过程中的低能构象,用信息熵控制最优解搜索空间的收缩,并用空间收缩的尺度作为算法停止的判据,进化过程中添加了最优保留策略,从而确保了算法的全局收敛性. 对于多约束优化问题(5),可利用评价约束函数PEC及精准惩罚函数法将其转化为序列无约束优化问题: 式中:α为惩罚因子,α只要大于一个阈值就可以使问题的解位于可行域内;ψ的取值一般为[103,105],这种方法针对所有约束按“松”与“紧”自动调整惩罚力度,能够有效地处理约束,计算效率较高.对于遗传算法,需要将上式转化为无约束最大化问题: 式中:C是一个大的正数以确保F(x)在计算过程中为正值,式(7)就是本文采用的演化设计模型,F(x)为适应值函数. 将通讯论中的信息熵理论引入优化方法中,构造基于信息熵控制的遗传演化模型如下: 式中:M为种群个数,通过定义最优解落在第m个种群的概率pm(m=1,2,…,M),从而引入信息熵H以衡量最优解落于某一种群的不确定性.初始时,pm=1/M,m=1,2,…,M,H取最大值;随着优化的进行,遗传迭代解将逐步逼近最优解,pm及H都将随之变化,当在某一种群取到最优解时,不确定性为零,熵 H取极小值,从而得到原问题(7)的最优解.信息熵的介入有助于加快进化过程. 在本文算法中,还将遗传算法中的交叉概率及变异概率作为设计变量参与优化,这种自适应策略,可以有效防止过早收敛问题的发生,同时提高了算法的搜索速度,保持了种群的多样性,从而大大降低了人为因素对优化算法的影响. 这种基于信息熵的多种群自适应遗传算法,引入了种群竞争机制及交叉、变异概率的自适应策略,并用信息熵控制空间收缩,提高了遗传迭代的效率,算法稳定可靠,具有较强的全局寻优能力,收敛速度也有较大的提高. 本文将知识打分函数与优化算法相结合,开发了新的分子对接程序.为测试程序的有效性,选取乙酰胆碱酯酶抑制剂(AChE)、凝血酶抑制剂(thrombin-MQPA)及HIV蛋白酶抑制剂3种晶体复合物,进行晶体结构复原,并与广泛应用的分子对接程序DOCK(Kuntz研究组推出的最新版本DOCK6.1)在能量得分、均方根偏差和对接所消耗的计算机时间方面进行了比较,得到了较为满意的结果. 凝血酶是与血液凝固有关的重要蛋白酶之一,它能水解L-精氨酸的肽、酰胺和酯类.临床表明,凝血酶抑制剂对血液栓塞、外伤出血等与血液凝固相关的疾病具有较好的疗效.本文选取凝血酶(PDB:1ETR)复合物中配体MQI与其受体进行分子对接,其对接结果如表3及图1所示,表中energy为能量得分,单位kJ/mol;RMSD为晶体结构的均方根偏差,单位nm;time为对接时间,单位s. 表3 1ETR:对接结果与DOCK 6.1的比较Tab.3 1ET R:Comparisons of the docking results with DOCK 6.1 老年痴呆症 (alzheimer′s disease,AD)是一种多因异质性疾病,伴有认知及行为障碍,多发生在65岁以上的老年人群.由于AD的病因病机尚未明确,目前对该病尚无特效药物.目前从血液和脑脊液中发现一些具有诊断和鉴别诊断意义的生化指标,有望成为AD早期诊断极有价值的指标,乙酰胆碱酯酶就是其中之一. 图1 1ET R:对接结果与晶体结构的比较Fig.1 1ETR:Comparisons of the docking results with crystal structure 本文运用改进的方法,对乙酰胆碱酯酶(PDB:1EVE)抑制剂晶体复合物中配体E20与其受体作对接,与DOCK6.1的对接结果相比较,结果如表4及图2所示. 表 4 1EVE:对接结果与 DOCK 6.1的比较Tab.4 1EVE:Comparison of the docking results with DOCK 6.1 人类免疫缺陷病毒(HIV)是艾滋病的主要致病因,针对艾滋病的化学药物治疗中HIV蛋白酶抑制剂发挥了重要作用.它通过抑制HIV在复制后期的构造蛋白、调节蛋白的功能,使其成为无外膜蛋白构造且不具感染力的病毒,从而达到治疗作用.本文选取 HIV蛋白酶中的一种(PDB:1QBS),将其与配体DMP进行对接,其晶体结构与对接结果如表5及图3所示. 图2 1EVE:对接的最优构象与晶体结构的比较Fig.2 1EVE:Comparisons of the optimal docking conformations with crystal structure 表5 1QBS:对接结果与DOCK6.1的比较Tab.5 1QBS:Comparison of docking results with DOCK6.1 图3 1QBS:对接的最优构象与晶体结构的比较Fig.3 1QBS:Comparisons of the optimal docking conformations with crystal structure 由上述3个实例可以看出,对于活性位点形成氢键或结合位点存在疏水性口袋的复合物(如1ETR、1QBS),本文的方法精度远好于DOCK6.1.这是由于本文采取的打分函数并不单纯以力场作为衡量能量的标准,而是通过将原子对间的距离分布转化为受体与配体分子间的结合能,从而将难以用公式显性表达的氢键、疏水等结合过程中的力隐含在概率分布中,因而得到了更好的结果.同时,3个实例均表明,本文的方法在保证精度的前提下,效率优于DOCK6.1的结果. 打分函数的选取与搜索算法的改进是分子对接过程中较为重要的两个部分.本文在传统对接程序DOCK的基础上,采用基于原子间概率分布的知识打分函数替代了基于力场的打分函数;同时采用基于信息熵的多种群自适应遗传算法,发展出一种新型对接程序KGAsDock,通过算例证明该方法在保证效率的前提下,提高了计算的精度,得到了较为满意的结果. [1]KUNTZ I D,BLANEY J M,OA TLEY S J,et al.A geometric approach to macromolecule-ligand interactions[J].Journal of Molecular Biology,1982,161(12):269-288 [2]EWING T J,M AKINO S,SKILLMAN A G,et al.DOCK4.0:Search strategies for automated molecular docking of flexible molecule databases[J].Journal of Computer-aidedMolecularDesign,2001,15(5):411-428 [3]LI Hong-lin,LI Chun-lian,GUI Chun-shan,et al.GAsDock:a new approach for rapid flexible docking based on an improved multi-population genetic algorithm[J].Bioorganic&Medicinal Chemistry Letters,2004,14(18):4671-4676 [4]SIPPL M J.Boltzmann′s principle,knowledge-based meanfields and protein folding.An approach to the computational determination ofprotein structures[J].Journal of Computer-aided Molecular Design,1993,7(4):473-501 [5]M UEGGE I.PMF scoring revisited[J].Journal of Medicinal Chemistry,2006,49(20):5895-5902 [6]李纯莲,王希诚,赵金城,等.一种基于信息熵的多种群遗传算法[J].大连理工大学学报,2004,44(4):589-593(LI Chun-lian,WANG Xi-cheng,ZHAO Jin-cheng,et al.An information entropy-based multi-population genetic algorithm[J].Journal of Dalian University of Technology,2004,44(4):589-593)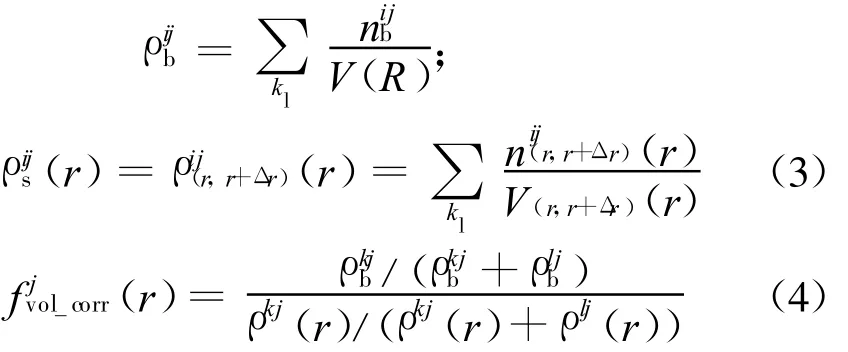
2 分子半柔性对接优化模型
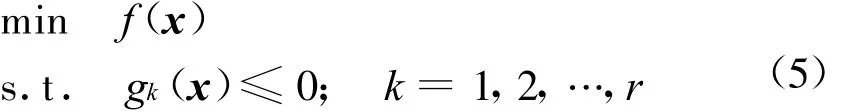
3 基于信息熵的多种群自适应遗传算法
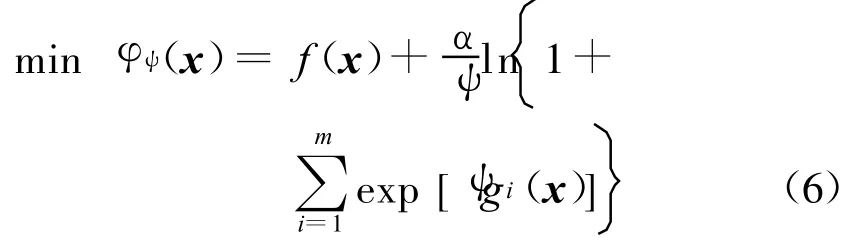
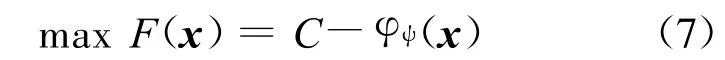
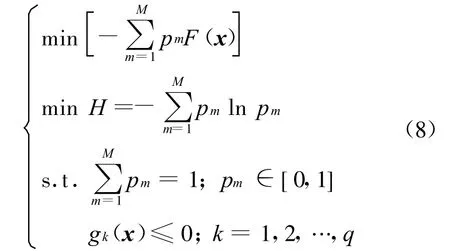
4 结果与讨论
4.1 凝血酶抑制剂晶体结构复原

4.2 乙酰胆碱酯酶抑制剂(AChE)晶体结构复原

4.3 HIV蛋白酶抑制剂晶体结构复原

5 结 语
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
雷达学报(2017年6期)2017-03-26
材料科学与工程学报(2016年4期)2017-01-15
衡阳师范学院学报(2016年3期)2016-07-10
合成化学(2015年4期)2016-01-17
池州学院学报(2015年3期)2016-01-05
火炸药学报(2014年3期)2014-03-20
郑州大学学报(理学版)(2014年2期)2014-03-01
无机化学学报(2014年6期)2014-02-28
