
过渡金属催化剂加氢脱硫作用机理研究
2010-09-11 11:48王爱华赵留喜刘君丽
河南化工 2010年19期
王爱华,赵留喜,刘君丽
(1.河南省化工研究所有限责任公司,河南郑州 450052;2.郑州工业贸易学校,河南郑州 450007)
过渡金属催化剂加氢脱硫作用机理研究
王爱华1,赵留喜2,刘君丽1
(1.河南省化工研究所有限责任公司,河南郑州 450052;2.郑州工业贸易学校,河南郑州 450007)
从过渡金属催化剂的活性相结构和反应物在催化剂表面活性位上的吸附—催化反应机理两个方面阐述了过渡金属催化剂的催化作用研究进展,并对过渡金属催化剂催化机理研究存在的争议和未来的研究方向进行了分析。
加氢脱硫;过渡金属催化剂;催化作用机理;活性相
Abstract:The research progress such as structure of the active phase and the mechanism of adsorptionto-reaction of reactants on the active sites at the surface of transition metal catalysts are reviewed.The controversies in the research of catalysis mechanism of transition metal and the direction in the future are also propounded.
Key words:hydrodesulfurization(HDS);transition metal catalyst;catalysis mechanism;active phase
1 引言
随着含硫原油加工量的增加以及重油催化裂化(FCC)技术的普及,轻质油含硫量超标及安定性差的现象变得较为突出。目前降低FCC汽油中硫含量的常用技术有催化加氢、催化氧化、分馏、碱液处理、再裂化重汽油等。加氢精制是石油加工的重要过程之一,主要是通过催化加氢脱除油品中的有害杂质(如S、N、O及金属等有机化合物),对二次加工后的柴油精制来说,还包括使烯烃、二烯烃、芳烃和稠环芳烃选择加氢饱和,并脱除金属等杂质,从而使油品能够更好地满足深加工的要求,达到资源的合理利用。由于原料结构及组成的差异,有些加氢精制过程可直接生产合格产品,而有的只能为下游工艺过程提供优质进料,如重整预加氢、加氢裂化一段精制及渣油加氢处理等过程[1]。由于原油重质化日益严重,开发和研制出具有高加氢脱硫活性的催化剂来满足油品深度加氢处理的需求成为重要任务[2]。
2 加氢脱硫反应机理
石油馏分中存在的有机硫化合物主要有硫醇、硫醚、二硫化物、噻吩、苯并噻吩(BT)、二苯并噻吩(DBT)及其衍生物等。对于噻吩、BT、DBT或其衍生物,其反应活性要低很多,特别是对于DBT及其衍生物类,它们是制约深度脱硫的重要因素;而对于更复杂的含硫稠环化合物,不仅脱硫极为困难,甚至其反应机理也还有待进一步研究。对于几种模型化合物在传统的加氢脱硫(HDS)催化剂上的反应活性一般具有如下的顺序:噻吩>苯并噻吩>苯萘并噻吩>四氢苯萘并噻吩>二苯并噻吩>4,6-二甲基二苯并噻吩。
2.1 噻吩加氢脱硫的反应机理
噻吩的转化存在两种途径:一是直接脱硫的历程,即表面活化的H使C—S键断裂;另一种是加氢脱硫的历程,即先经过加氢饱和再通过氢解而除去S,研究表明要使芳环中的C—S直接断裂是相当困难的。对于噻吩的加氢脱硫,认为先进行的氢化饱和,然后再进行C—S的断裂,由于加氢饱和破坏了噻吩环的芳香性,使得脱硫变得容易。在这一过程中二氢噻吩的氢解与C==C的加氢存在着竞争;但由于噻吩、二氢噻吩和四氢噻吩的加氢脱硫具有相同的产物分布,因而认为是按照相同的机理进行的。在Mo(110)单晶表面进行的吸附研究表明,噻吩、四氢噻吩和2,5-二氢噻吩的反应机理是不同的。噻吩和四氢噻吩可能是通过2,5-二氢噻吩中间物来完成脱硫的,而硫醇盐则可能是按照完全不同的机理进行的[3]。
2.2 BT的加氢脱硫机理
BT在进行加氢脱硫时作为产物只发现乙苯和少量的二氢苯并噻吩,而二氢苯并噻吩进行加氢脱硫时,并没有观察到BT的形成。于是认为乙苯是通过二氢苯并噻吩作为中间物所形成的。于是,VanPanijs等[4]提出了一个如图1所示的平行反应历程。与噻吩的加氢脱硫相似,也有直接脱硫和加氢脱硫两条途径。
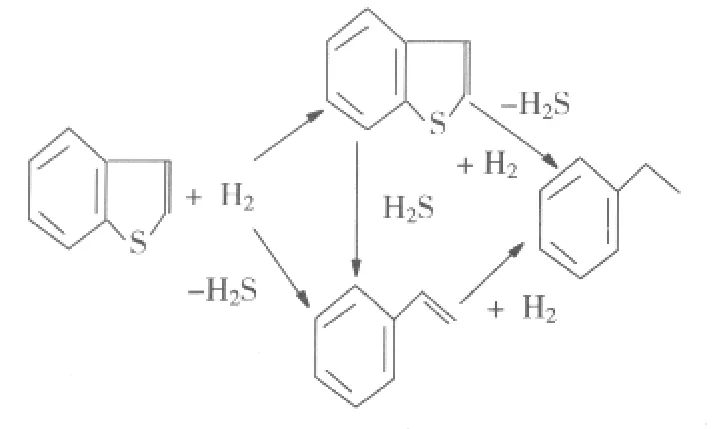
图1 BT加氢脱硫的反应途径
2.3 DBT的加氢脱硫机理
对于DBT的加氢脱硫,Borderick D等[5]提出了图2所示的DBT加氢脱硫的连续反应机理,因为发现联苯(Bi-Ph)是反应的主要产物,而环己基苯(CHB)只有极少量,在添加Bi-Ph和H2S的条件下进行DBT的加氢脱硫时,发现Bi-Ph的添加显著地减少了DBT的转化率,而添加H2S则没有影响,这意味着DBT的加氢脱硫受到了Bi-Ph的抑制。Houalla等[6]则指出反应可以通过最少量的氢耗来完成,且Bi-Ph与CHB的加氢速率很低。DBT的加氢速率随着H2S的浓度的增加而增加,并且还取决于催化剂的组成。当采用NiMo/Al2O3催化剂时得到的CHB的浓度比采用相似的CoMo/Al2O3催化剂高两倍。进一步研究表明,氮化物在催化剂表面的吸附比硫化物(从动力学的角度来说)更为有利,也不易脱附。氮化物的存在对反应物环上加氢的影响比直接脱硫的影响更大。在加氢处理过程中,氮化物中的N是以NH3的形式被脱除;因此,在工业上对循环气体进行净化于加氢脱硫也是有利的。
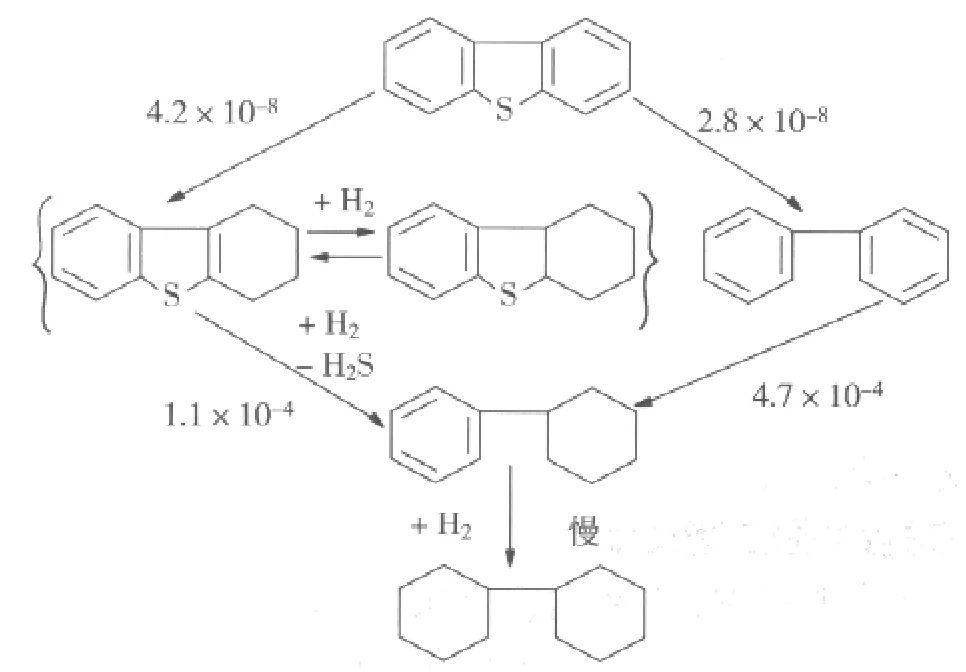
图2 DBT加氢脱硫反应途径
3 加氢脱硫催化剂
加氢精制催化剂一般以ⅥB族金属为活性组分(如Mo和W),以Ⅷ族金属为助催化剂(如Co和Ni),通常都是一些载体型复合金属氧化物或硫化物等;其中,MoCo/TiO2-Al2O3性能较优,已经普遍在工业中应用。对于以Co和Ni为助剂的MoO3和WO3催化剂在加氢脱硫活性方面的差异,研究结果认为Co-Mo、Ni-Mo和Ni-W几种催化剂的加氢脱硫活性依次递减。
近年来,利用过渡金属氧化物制备高比表面积催化新材料——过渡金属氮化物/碳化物和磷化物催化剂的研究已引起极大关注。在石油加氢精制反应中,负载型Mo2N作为噻吩加氢脱硫催化剂,其催化活性是Mo2S/Ai2O3催化剂的1.1~1.2倍,与硫化钼催化剂相比,氮化钼催化剂有较好的加氢脱硫活性和选择C—S键断裂的选择性;但负载型Mo2N催化剂的研究多以Al2O3为载体。金属氮化物催化剂通常具有优异的加氢脱氮(HDN)性能,但在油品加氢精制过程中则要求兼具加氢脱硫和HDN两种功能。以TiO2为载体的新型加氢脱硫催化剂还具有低温高活性的优势。
3.1 过渡金属硫化物催化体系
传统的过渡金属硫化物催化体系是由氧化铝担载的钴和钼的氧化物所组成,故通常被称为钼酸钴催化剂。该催化体系硫化之后虽然比贵金属的加氢活性低,但其所具有的抗硫性能使其得以广泛应用。经过多年的研究,已开发出多种性能优异的工业催化剂。
对于过渡金属硫化物催化剂加氢脱硫和HDN的催化作用机理还存在很多争议,大多数研究者普遍接受Topsóe等[7]提出的Co-Mo-S或Ni-Mo-S活性相理论,并认同在Co-Mo和Ni-Mo-S硫化物表面存在S阴离子空穴和解离吸附的H2分子形成的—SH基,有机硫化物分子通过S原子在S空穴上的“端连吸附”是氢解脱硫(直接脱硫)的重要途径[8]。关于Co-Mo-S和Ni-Mo-S活性相的存在只是得到了EXAFS(广延X射线吸收精细结构谱)和MOssbauer(穆斯堡尔)谱等方面不太确凿的证据,关于S空穴和—SH基的存在也只是得到了35S示踪实验单方面的间接证据。
关于有机硫化物在金属硫化物催化剂表面的吸附状态,大多数研究者普遍接受存在通过S原子的“端连吸附”和通过芳香环(或噻吩环)上的π电子的“平躺吸附”的观点[9]。但是,“端连吸附”和“平躺吸附”状态的假设只是得到了立体位阻的4-甲基二苯并噻吩(4-MDBT)和4,6-DMDBT(4,6-二甲基二苯并噻吩)加氢脱硫活性(反应速率常数)和二苯并噻吩(DBT)加氢脱硫活性的显著差异的间接证明,并未得到更强有力的直接证据。
3.2 过渡金属氮化物/碳化物
过渡金属氮化物/碳化物是一类金属间充型化合物,其表面性质和催化性能类似于Pt和Rh等贵金属元素,被誉为“准铂催化剂”,具有优异的加氢性能,作为一种新型的加氢精制催化新材料已引起广泛关注。
Me Crea等[10]通过H2-TPD研究发现,新鲜Mo2N催化剂或钝化Mo2N催化剂还原后在室温对氢显示出较强的吸附活性,生成了不可逆吸附氢。这些室温生成的不可逆吸附氢主要在573~773 K温区脱附,说明生成了强吸附的氢物种。依照异裂机理,吸附在催化剂表面的H2被活化生成。由于键键能较低易断键,氢以活化态物种的形式从催化剂表面转移到反应物分子上;同时,带一个负电荷的氢也会转移到次表面或晶隙中。这主要是因为在次表面上晶体中的N和C元素占支配地位,而N—H键和C—H键的键能大于Mo—H键的键能[11];另外是因为氮化物/碳化物晶格中残余氧的存在所致。O—H键的键能比C—H键和N—H键还要强,这也促使表面氢向次表面移动,结果使表面氢和次表面氢之间达到一个平衡状态。因此,在加氢过程中次表面的作用就像一个储氢容器,对催化剂活性表面的氢进行转移和补充。众多研究表明,氮化钼催化剂的加氢脱硫活性高于传统的硫化钼催化剂,具有较好的工业应用前景。
3.3 过渡金属磷化物催化剂体系
有关磷化物催化活性的研究报道不多,最近几年才开始有关磷化物催化剂在加氢脱硫和HDN方面的研究。过渡金属磷化物受到人们极大关注的一个主要原因是,因为磷化物比氮化物和碳化物具有更好的抗硫中毒性能。与硫化物催化剂相比,磷化物催化剂具有较高的加氢脱硫反应活性。
Phillips等[12]的研究发现,在噻吩加氢脱硫反应中,反应进行150 h,15%MoP/SiO2催化剂的活性是相同金属担载量的MoS2/SiO2的4倍。Oyama[13]比较了不同磷化物在模型原料(喹啉、二苯并噻吩和四氢萘的十四烷混合溶液)中的加氢脱硫活性,这些磷化物的活性顺序是Fe2P<CoP<MoP<WP<Ni2P。
图3为磷化物催化剂的DBT加氢脱硫反应机理示意图。DBT反应产物是联苯(BP)、环己基苯(CHB)和四氢二苯并噻吩(H4-DBT)。DBT脱硫主要通过两条平行的路线,即:①直接脱硫(DDS)得到BP,然后BP再进一步加氢,得到CHB;②加氢之后再脱硫(HYD),首先得到H4-DBT,然后进一步加氢得到CHB。
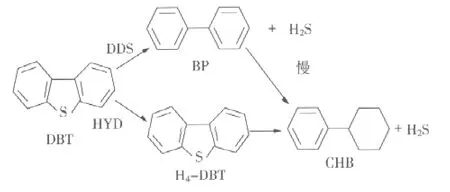
图3 磷化物催化剂的DBT加氢脱硫反应机理示意图
从DBT加氢脱硫反应活性来看,很明显,采用DDS路线得到的BP是反应的主要产物,它的选择性占65%。通过BP加氢和H4-DBT脱硫可以得到CHB,但不论哪种路线生成CHB,它的选择性都较低,中间产物H4-DBT的选择性最低,少于10%。由此可以得出结论,对于磷化物催化剂而言,DBT脱硫主要采用直接脱硫(DDS)的路线,可以使主要产物是BP而不是CHB。这是所希望的,因为这样可以减少HDS过程中的氢耗和避免烃类结构的裂解。
噻吩和二苯并噻吩的HDS反应结果表明,在HDS反应过程中,磷化物催化剂的结构保持不变,仅表面被部分地硫化,很可能生成了磷硫活性相。深度硫化的磷化物,在比较温和的条件下可以被完全活化到新鲜的磷化物的状态。过渡金属磷化物MoP/SiO2、Ni2P/SiO2和NiMoP/SiO2催化剂的DBT HDS反应活性顺序是:Ni2P/SiO2>NiMoP/SiO2>MoP/SiO2。对于MoP、Ni2P和NiMoP催化剂,DBT加氢脱硫反应主要采取C—S键断裂的直接脱硫途径,反应主要产物是联苯。Ni2P/SiO2催化剂所具有的优异而稳定的催化活性的原因很可能是在Ni2P/SiO2催化剂的表面生成了NiPxSy活性相。
4 结束语
尽管不同模型对催化剂活性相的解释有所不同,但这些模型也有共同点,即加氢脱硫反应活性中心与催化剂表面存在的配位不饱和阳离子中心(即阴离子空位)密切相关。
由于金属催化剂(尤其是过渡金属硫化物)对反应物(H2、有机硫化物、氮化物、烯烃和芳烃等)的吸附活性低,表面吸附物种浓度小,因此金属催化剂上加氢脱硫的催化作用机理研究十分困难。况且,金属催化剂只有在加氢反应条件下才具有催化活性,由于过去缺乏“原位”反应检测和分析手段,很难得到催化剂在反应状态的结构和表面反应物种的直接证据。虽然通过STM技术和DFT计算对金属催化剂活性相的研究取得了很好的结果,但是,由于STM实验是以金(Au)作为载体基质,以金属Mo和Co作为MoS2和Co-Mo-S的前驱物,而工业催化剂一般是以Al2O3作为载体,以MoO3和CoO作为MoS2和Co-Mo-S前驱物,涉及到金属氧化物在载体Al2O3上的硫化过程,所以当前STM的研究结果具有一定的局限性。
随着现代仪器分析技术的发展,尤其是原位STM、原位FT2IR、原位XRD、近似原位XPS、近似原位EXAFS、动态微反—色谱、TPR-MS、TPD-MS和TPS-MS等技术的应用,使我们在反应状态下“零距离”直接检测和分析金属硫化物催化活性相的结构,表面吸附物种和表面反应物种成为可能。
[1]廖士纲,韩崇仁.面向21世纪的加氢裂化技术[J].炼油设计,1999,29(6):1-3.
[2]鹿清华,张国生,何诈云.高硫减压渣油加工的经济性研究[J].当代石油化工,2003,11(3):7-9.
[3]朱全力,赵旭涛,赵振兴,等.加氢脱硫催化剂与反应机理的研究进展[J].分子催化,2006,20(4):372-383.
[4]Vanparijs I A,Froment G F.Advances in Hydrodesulfurization Mechanism and Catalysts[J].Ind Eng Chem Res.1986,25:431-442.
[5]Broderick D H,Sapre A V,Gates B C.Study on the reaction mechanism of 4,6-dimethyldibenzothiophene hydrodesulphurization on Co-Mo/γ-Al2O3catalyst[J].Am Chem Soc Div Petro Chem Pro,1977,22:941-942.
[6]Houalla M,Broderick D H,Sapre A V,et al.Research development of kinetics for hydrodesulfurization catalytic process[J].Catal,1980,61:523.
[7]Clausen B S,Moerup S,Topsóe H,et al.The research on FCC gasoline deep desulfurization technology[J].Physique,1976,37:249-257.
[8]Kabe T,Qian W H,Ogawa S,et al.Mechanism of the adsorption of metal lo-porphyrin of the surface of HDS[J].Catal,1993,143:239-248.
[9]柴永明,安高军,柳云骐等.过渡金属硫化物催化加氢作用机理[J].化学进展,2007,19(2/3):234-242.
[10]McCrea K R,Logan J W,Tarbuck T L,et al.Progress in thiophenic sulfide compond catalytic hydrodesulfurization[J].Catal,1997,171:255-258.
[11]Colling C W,Thompson L T.Reasearch progress in deep hydrodesulfurization by molybdenum nitride catalyst[J].Catal,1996,146(1):193-203.
[12]Phillip S D C,Sawhill S J,Self R,et al.HDS,HDN and HDO of FCC gasoline spiked with benzothiophene over PtPd/H-USY[J].Catal,2002,207(2):266-273.
[13]Oyama S T.Property-reactivity correlation for HDS of middle distillates[J].Catal,2003,216(1-2):343-352.
Study on Hydrodesulfurization Mechanism of Transition Metal Catalyst
WANG Ai-hua1,ZHAO Liu-xi2,LIU Jun-li1
(1.Henan Chemical Industry Research Institute,Zhengzhou 450052,China;2.Zhengzhou Trade and Industry School,Zhengzhou 450007,China)
TQ426.8
A
1003-3467(2010)19-0034-04
2010-05-21
王爱华(1979-),女,硕士,主要从事液相催化加氢研究工作,电话:13663004620。
猜你喜欢
电镀与精饰(2022年10期)2022-10-14
电镀与精饰(2022年3期)2022-03-14
石油炼制与化工(2021年5期)2021-05-12
航空制造技术(2020年15期)2020-11-06
表面工程与再制造(2019年6期)2019-08-24
表面技术(2019年6期)2019-06-26
石油化工腐蚀与防护(2017年1期)2017-08-15
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
