
NaAlH4表面Ti催化空间构型和X射线吸收光谱: Car-Parrinello分子动力学和密度泛函理论研究*
2010-09-08 06:06周晶晶陈云贵吴朝玲肖艳高涛
物理学报 2010年10期
周晶晶陈云贵†吴朝玲肖艳高涛
1)(四川大学材料科学与工程学院,成都610065)
2)(四川大学原子与分子物理研究所,成都610065)
(2009年12月1日收到;2010年1月5日收到修改稿)
NaAlH4表面Ti催化空间构型和X射线吸收光谱: Car-Parrinello分子动力学和密度泛函理论研究*
周晶晶1)陈云贵1)†吴朝玲1)肖艳1)高涛2)
1)(四川大学材料科学与工程学院,成都610065)
2)(四川大学原子与分子物理研究所,成都610065)
(2009年12月1日收到;2010年1月5日收到修改稿)
通过采用Car-Parrinello分子动力学方法对掺杂Ti前后的NaAlH4(001)2×2×1超晶胞表面晶体在333 K (60℃)温度条件催化脱氢的空间构型做了理论研究,发现掺杂Ti的合金中AlH4团的其中两个Al—H键长分别从约1.64(1=0.1nm)增大至1.74和1.93,而未掺杂合金表面中AlH4团的4个Al—H键长基本不变,这意味着掺杂Ti相对未掺杂的合金更易于放氢.但在模拟温度条件下并未发现Ti-Al成键趋势,分析认为这可能在于模拟过程温度不够高.在所得表面晶体结构基础上,采用全势能线性缀加平面波方法计算了TiAl3,TiH2晶体及Na8Ti8Al16H64(001)表面晶体的Ti K边X射线近边结构(XANES)谱,通过与实验测试的XANES和扩展X射线吸收精细结构对比发现,Ti原子不仅可能是以TiAl3和TiH2混合物形态存在,一部分Ti还可能取代NaAlH4表面的Na原子而存在.
NaAlH4,Car-Parrinello分子动力学,密度泛函理论,X射线吸收近边结构
PACC:8640K,7115Q,7115M,7870D
1. 引言
随着化石燃料能源危机加重和生态环境恶化,在未来社会中氢能无疑是最具潜力的清洁新能源之一[1,2].其实用化最具挑战的技术在于储氢,大容量安全存储、适宜的工作条件、长寿命的循环次数等均是其重要性能指标.NaAlH4的大容量储氢特性一经提出就引起了研究者的广泛关注,并有大量关于TiCl3催化脱氢技术的研究报道,特别是其催化微观机理方面.美国标准与技术研究所(NIST)[3—5]采用第一性原理模拟计算与中子非弹性散射相结合的研究方法,得知催化剂TiCl3中的Ti原子优先取代了NaAlH4合金中的Na原子,从而弱化了Al—H的键能进而使得合金更易吸放氢.在合金表面也有类似情况,吸附的H分子更易分解成H原子.另有分子动力学的模拟研究表明TiAln(n>1)附近有氢富集[4],这也有利于吸氢.这些理论研究结果与实验上核磁共振(NMR)[6,7]、X射线衍射(XRD)[6,7]以及X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)[8—10]测试分析相一致.XANES和EXAFS是研究催化剂与反应物形态的非常有效的检测手段[11,12].理论上可以通过建立Ti取代Na占据晶格位置的模型,采用第一性原理计算得到XANES谱[13],最后对比理论假设模型的XANES计算谱和实验谱,就可以推断假设模型的存在与否.但是,仅凭实验难以确定Ti在NaAlH4合金表面的催化微观模型.为此,本文先采用Car-Parrinello分子动力学(CPMD)确定TiCl3在NaAlH4合金表面催化作用的结构形貌,再进一步采用第一性原理全势能线性缀加平面波(FPLAPW)方法计算表面合金的中掺杂Ti K边XANES,分析Ti在NaAlH4合金表面的微观形态和作用机理.
2. 计算模型与方法
2.1. 模型建立
NaAlH4单胞结构的晶格常数为a=b=4.98(1=0.1nm),c=11.05,空间群I41/a,Na,Al和H原子分别占据4a(0,1/4,1/8),4b(0,1/4,5/ 8)和16f(0.2335,0.3918,0.5439)晶格位置.根据已报道的研究结果,催化剂TiCl3中的Ti原子优先取代NaAlH4合金表面的Na原子,并且还可知催化剂在(001)和(100)表面催化作用没有任何区别[4].因此,我们仅选择了(001)表面作为研究对象.为了实现微量掺杂及提高XANES计算准确性,特建立了2×2×1的超晶胞结构.另外,在(001)表面外添加了12真空层,还将靠近真空层的4个Na原子中的两个用Ti原子取代.最终所得表面晶体结构可表示为Na8Ti8Al16H64(001)(如图1所示).
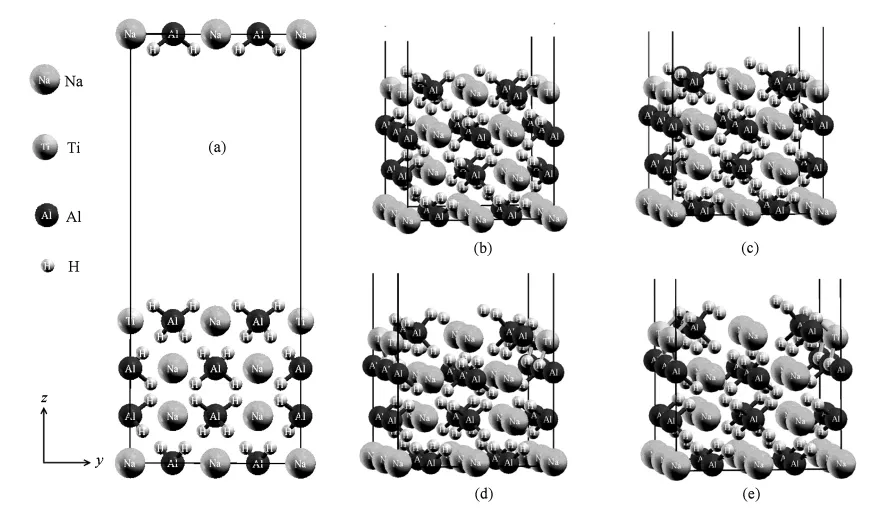
图1 Na8Ti8Al16H64(001)表面晶体结构初始结构(a),经过2,10,20和30 ps动力学模拟后的结构分别为(b),(c),(d)和(e)
在XANES谱计算时,为了对比分析,一并计算了TiAl3和TiH2的XANES谱.其中,TiAl3的晶体结构常数为:a=b=3.841,c=8.639,空间群I4/mmm;而TiH2的晶体结构常数为:a=b=c= 4.431,空间群Fm-3m.为保证XANES计算准确性和可比较性,对TiAl3和TiH2也同样构建了2×2×1的超晶胞结构.
2.2. 计算方法
2.2.1. 结构优化
为确定特定温度下合金表面晶体结构,本文采用了CPMD方法[14]优化晶体结构,相应程序是CPMD3.11[15].计算程序采用赝势平面波方法,在周期性边界条件下,平面波截断能设定为400 eV,体系中的所有原子的芯电子与价电子关系均采用Vanderbilt超软赝势[16,17]描述,计算中还采用了Perdew-Burke-Ernzerh(PBE)泛函形式的广义梯度近似(GGA)[18].此外,本文选用了Verlet运算法则[19]求解Newton运动方程.研究体系在333 K(60℃)温度的正则系综(NVT)下,采用Nosé-Hoover调温水浴[20,21],经历了30000步步进为1 fs的动力学模拟,特征频率取1000cm-1.另外,虚拟电子质量为450 a.u.,原子核质量分别是:H原子1.0 amu,Al原子27.0 amu,Na原子23.0 amu和Ti原子47.9 amu.
需要说明的是,在模拟中,为接近真实环境防止底层原子运动到真空层,特束缚了靠近底层的两层原子;为防止过渡态阻碍导致局部弛豫不均,在模拟前期先采用了400K高温1500cm-1高频弛豫表面晶体结构.
2.2.2. XANES谱计算
XANES谱计算采用WIEN2K-08程序,该程序是以密度泛函理论(DFT)为基础的第一性原理的全电子计算方法——FPLAPW方法[22,23].该方法通过引入线性组合的球谐函数和平面波基函数求解Kohn-Sham方程,基函数的选取以及对Kohn-Sham方程自洽场求解均基于muffin-tin模型,其中各原子muffin-tin半径取值分别为:Rmt(Na)=1.8 a.u.,Rmt(Ti)=2.0 a.u.,Rmt(Al)=1.6 a.u.和Rmt(H)= 1.4 a.u.,且取Rmt×Kmax=3.5 Ry来确定平面波截断能(Kmax为最大倒格矢),并用-6.0 Ry分开芯电子和价电子层.另外,不可约Brillouin区取K点为13×6 ×2,电子的交换关联能采用了Wu-Cohen06[24]GGA方法,自洽迭代求解的收敛精度为0.0001 Ry.
Ti K边XANES计算谱对应掺杂Ti原子的s轨道上一个电子被激发为自由电子的吸收谱,谱线展宽采用了Lorentz函数拟合方式,时间展宽因子取2,光谱展宽因子取1.0,为了比较展宽效果,对TiH2的计算还多取展宽因子1.5.
3. 结果与讨论
3.1. 结构分析
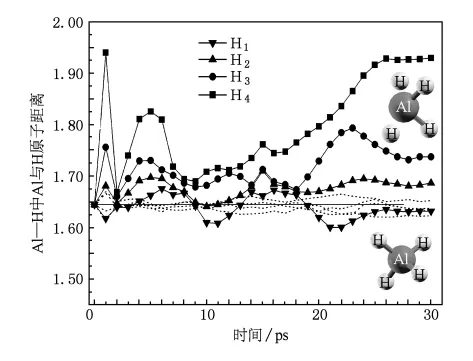
图2 Na8Ti8Al16H64(实线加符号)和Na16Al16H64(虚线)(001)表面晶体中AlH4团的Al—H原键中Al与H子间距随模拟时间变化
对Na8Ti8Al16H64(001)表面晶体结构经过30 ps动力学模拟,其中第2,10,20和30 ps时的结构如图1(b)—(e)所示.可以发现,Na8Ti8Al16H64(001)表面晶体结构中的AlH4团周围的H原子有远离Al原子的趋势,即AlH4团趋于分解.为了定量分析Al—H键中Al和H原子间距随时间的变化情况,将初始结构时间定义为0 ps,并随同后面30 ps动力学模拟所得合金表面晶体结构中AlH4团的Al与H原子间距做统计分析,将每皮秒末的动力学模拟结构加以比较,如图2所示.显然,AlH4团中只有1个H原子靠近Al原子,其他3个H原子都有不同程度的远离趋势,从最初的1.64分别增大至1.69,1.74和1.93.在最后优化所得到的AlH4团如图2上方的原子团结构所示,很明显有两个H原子已无法与Al原子成键了,此即NaAlH4→1/3Na3AlH6+ 2/3Al+H2反应中释放的一个H2分子.对比分析未掺杂Ti原子的Na16Al16H64(001)表面晶体结构的动力学模拟数据,因模拟温度低于纯NaAlH4的放氢温度,故发现此结构中Al—H键中Al和H原子间距仅在平衡间距附近振动,并无很大的分离现象,即无放氢趋势,优化所得结构如图2下方的原子团结构所示.由此可见,TiCl3中Ti原子取代NaAlH4表面的Na原子可以弱化AlH4团中Al—H的键能,从而使得合金更有利于放氢.通过图3中Ti原子与其最近邻的Al和H原子间距变化,可发现经25 ps模拟后,掺杂Ti的NaAlH4表面中Ti-Al和Ti-H原子间距均与最初2.82301基本相等,且处于Chaudhuri等估计的2.7—2.9范围内[25].但是Ti-Al原子间距并没有达到TiAl3团生成成键间距2.80[26],而且整个系统也已经趋于稳定状态.所以,基本可以排除因为反应生成此两产物的时间大于30 ps,即超出于模拟时间尺度的可能性.另外,文献[4]中Born-Oppenheimer分子动力学(BOMD)模拟300K温度条件下的Ti-Al原子间距实际也并非如研究者所提的1.75,通过曲线取数字后发现应该大约在2.85左右.模拟中未发现Ti和Al成键,其原因一方面可能是Ti和Al成键在第二步放氢过程1/3Na3AlH6→NaH+1/3Al+1/2H2中更易形成;另一方面可能是模拟的温度介于掺杂和未掺杂Ti的NaAlH4放氢温度之间,未达到TiAl3的生成温度(225℃—475℃)[9],而实验采用的球磨合成方式较易于生成TiAl3.后者可能性较大,微观上可解释为Ti原子没有足够能量经间隙过渡与Al原子成键,Liu等[27]曾利用DFT方法在NaAlH4间隙掺杂Ti的模型模拟得到了2.6的Ti-Al原子间距.
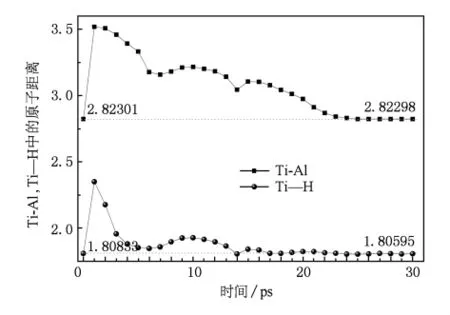
图3 Na8Ti8Al16H64(001)表面晶体中Ti原子与其最近邻Al和H原子的原子间距随模拟时间变化
3.2. XANES分析
为了确定Ti原子在晶体空间存在的构型以及是否有TiAl3和TiH2生成,我们计算了Na8Ti8Al16H64(001)表面晶体、TiH2和TiAl3晶体的XANES谱,并与实验测试结果对比分析.为此,将以上化合物的理论计算和实验测试的Ti K边XANES谱综合绘于图4.对比图4(a)TiH2和图4(b)TiAl3的Ti K边XANES计算谱和实验谱,可知计算谱和实验谱匹配较好.对比TiH2的两条不同展宽因子的XANES计算谱,发现展宽因子1.5拟合谱线较展宽因子1.0的拟合谱线更接近实验谱,但是同时也掩盖了一些特征峰信息.为了清晰地分析特征峰位,其他合金模型的XANES计算的光谱展宽因子均取值为1.0.

图4 Ti K边XANES理论计算和实验测试谱(a)TiH2实验谱(圆圈)[8]和不同光谱展宽因子1.0(虚线)和1.5(实线)下的两条计算谱;(b)TiAl3理论计算和实验测试谱(圆圈)[8]的对比;(c)掺杂Ti的NaAlH4氢化脱氢状态和TiAl3实验谱对比(上方3条实线)[8],以及纯TiAl3(下方实线)和掺杂20%TiH2(虚线)的试样实验测试结果[10];(d)Na8Ti8Al16H64(001)表面理论计算(实线)与掺杂Ti的NaAlH4实验测试谱(虚线)[8]对比
文献[8]比较了掺杂Ti的NaAlH4的脱氢前后(图4(c)上方两条谱线)、TiAl3(图4(c)第3条谱线)和TiH2(图4(a)中圆圈)的Ti K边XANES谱线,研究者认为掺杂Ti以TiAl3合金形式存在的可能性比较大.但是,结合图4(b)TiAl3的Ti K边XANES计算谱对比图4(c)中掺杂Ti的NaAlH4和TiAl3实验测试谱,发现掺杂Ti的NaAlH4的Ti K边XANES并没有TiAl3中4970.92与4977.72 eV明显的特征峰.Ignatov等[10]随后以8∶2的比列混合TiAl3和TiH2并测Ti K边XANES,发现混合物的Ti K边XANES谱线与掺杂Ti的NaAlH4的更接近.为此,Ignatov等推断TiCl3催化剂中的Ti原子是以TiAl3和TiH2混合物形态存在.
对比图4(d)中NaAlH4表面掺杂Ti后(即Na8Ti8Al16H64(001)表面)的Ti K边XANES计算谱与实验测试谱[8],二者在4975—4975 eV范围有明显差异,但二者在4990.74 eV附近的特征峰几乎完全重合,NaAlH4表面掺杂Ti模型的理论计算XANES谱比TiH2的特征尖峰(4980.72 eV)更接近掺杂TiNaAlH4的实验谱特征峰.所以,Ignatov等推断的混合物中,掺杂Ti原子取代NaAlH4(001)表面Na原子位置存在的可能性比以TiH2化合物形态存在可能性更大.所以,文献[10]推断的Ti原子是以TiAl3和TiH2混合物形态存在并不一定准确,还有可能一部分Ti原子以取代NaAlH4表面的Na原子存在.
4. 结论
通过对掺杂Ti的NaAlH4(001)表面采用CPMD方法优化其333 K温度下的空间构型,并采用FPLAPW全电子方法计算了掺杂Ti原子的K边XANES,同时还对比计算了Ti原子其他两种可能存在的化合物TiAl3和TiH2的Ti K边XANES.研究发现,掺杂Ti原子取代NaAlH4表面的Na原子可以弱化AlH4团中Al—H的键能,并分解了其中的两个H原子,对比未掺杂合金中AlH4团,发现掺杂后的合金更有利于放氢.333 K温度下模拟所得到的Ti-Al原子间距约为2.823,并无键形成,这可能是由于模拟温度过低无法克服过渡态激活能.结合计算的TiAl3,TiH2及Na8Ti8Al16H64(001)表面的Ti K边XANES谱和实验测试的XANES和EXAFS分析发现,Ignatov等推断的Ti原子是以TiAl3和TiH2混合物形态存在并不一定准确,也有可能一部分Ti原子以取代NaAlH4表面的Na原子存在.
[1]Wirth T E,Gray C B,Podesta J D 2003 Foreign Aff.82 132
[2]Kerr R A,Service R F 2005 Science 309 101
[6]Herberg J L,Maxwell R S,Majzoub E H 2006 J.Alloys Compd. 417 39
[7]Majzoub E H,Herberg J L,Stumpf R,Spangler S,Maxwell R S 2005 J.Alloys Compd.394 265
[8]Graetz J,Reilly J J,Johnson J,Ignatov A Y,Tyson T A 2004 Appl.Phys.Lett.85 500
[9]Baldé C P,Stil H A,van der Eerden A M J,de Jong K P,Bitter J H 2007 J.Phys.Chem.C 111 2797
[10]Ignatov A Y,Graetz J,Chaudhuri S,Salguero T T,Vajo J J,Meyer M S,Pinkerton F E,Tyson T A 2007 AIP Conference Proceedings 882 613
[11]Tang Y H,Lin L W,Guo C 2006 Aata Phys.Sin.55 4197(in Chinese)[唐元洪、林良武、郭池2006物理学报55 4197]
[12]Cao H B,Chen D L,He L H,Zhang J R,Wang F W,Wu Z Y,Yan Q W 2007 Chin.Phys.16 784
[13]Ma C Y,Cui M Q,Zhang L Y,Wu X,Zhou K J,Wu Z Y, Chen X,Zhao Y D,Zheng L 2008 Aata Phys.Sin.57 3868(in Chinese)[马陈燕、崔明启、张凌云、巫翔、周克瑾、吴自玉、陈兴、赵屹东、郑雷2008物理学报57 3868]
[14]Car R,Parrinello M 1985 Phys.Rev.Lett.55 2471
[15]Payne M C,Teter M P,Allan D C,Arias T A,Joannopoulos J D 1992 Rev.Mod.Phys.64 1045
[16]Vanderbilt D 1990 Phys.Rev.B 41 7892
[17]Laasonen K,Pasquarello A,Car R,Lee C,Vanderbilt D 1993 Phys.Rev.B 47 10142
[18]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[19]Verlet L 1967 Phys.Rev.159 98
[20]Nosé S 1984 Molec.Phys.52 255
[21]Hoover W G 1985 Phys.Rev.A 31 1695
[22]Blaha P,Schwarz K,Sorantin P,Trickey S B 1990 Compupt. Phys.Commun.59 399
[23]Schwarz K,Blaha P,Madsen G K H 2002 Compupt.Phys. Commun.147 71
[24]Wu Z,Cohen R E 2006 Phys.Rev.B 73 235116
[25]Chaudhuri S,Muckerman J T 2005 Phys.Chem.B 109 6952
[26]Colinet C,Pasturel A 2002 J.Phys.:Condens.Matter 14 6713
[27]Liu J,Ge Q 2006 Chem.Commun.17 1822
PACC:8640K,7115Q,7115M,7870D
*Project supported by the National High Technology Research and Development Program of China(Grant No.2007AA05Z114).
†Corresponding author.E-mail:ygchen60@yahoo.com.cn
Spatial configurations and x ray absorption of Ti catalyzing on NaAlH4surfaces:Car-Parrinello molecular dynamics and density functional theory study*
Zhou Jing-Jing1)Chen Yun-Gui1)†Wu Chao-Ling1)Xiao Yan1)Gao Tao2)
1)(School of Materials Science and Engineering,Sichuan University,Chengdu610065,China)
2)(Institute of Atomic and Molecular Physics,Sichuan University,Chengdu610065,China)
(Received 1 December 2009;revised manuscript received 15 January 2010)
A theoretical study on the spatial configurations of the catalytic dehydrogenation of the pre-and post-Ti-doped NaAlH4(001)2×2×1 supercell surface crystals was performed by using the Car-Parrinello molecular dynamics(CPMD) method at 333 K(60℃).It was be found that two of the Al—H bond lengths increased from approximately 1.64 to 1.74 and 1.93respectively in the AlH4groups of the Ti-doped alloy.Compared with this change,the four Al—H bond lengths almost kept invariant in the AlH4group of un-doped alloy,which means that it was easier to dehydrogenate for the Ti-doped alloy than un-doped alloy.There was no bonding tendency between atom Ti and Al observed,which is probably because the temperature in the simulation process is not high enough.Based on the obtained surface crystal configuration,the Ti K-edge x ray absorption near-edge structure(XANES)spectra of the TiAl3,TiH2crystals and Na8Ti8Al16H64(001) surface crystal have been calculated by using the full-potential linearized augmented plane wave method(FPLAPW).It was also found that the atom Ti may not only exist in the mixture of TiAl3and TiH2but also probably partially substitute for the Na atoms in NaAlH4surface crystal,by comparing the experimental XANES and edge x ray absorption fine structure (EXAFS)spectra.
NaAlH4,Car-Parrinello ab initio molecular dynamics,density functional theory,x ray absorption near-edge structure
book=613,ebook=613
*国家高技术研究发展计划(批准号:2007AA05Z114)资助的课题.
†通讯联系人.E-mail:ygchen60@yahoo.com.cn
猜你喜欢
机电安全(2022年5期)2022-12-13
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
科学(2020年1期)2020-01-06
中国交通信息化(2019年4期)2019-07-13
衡阳师范学院学报(2016年3期)2016-07-10
小学生导刊(低年级)(2016年5期)2016-05-27
中国卫生(2015年12期)2015-11-10
中国卫生(2015年10期)2015-11-10
火炸药学报(2014年3期)2014-03-20
印制电路信息(2014年11期)2014-03-11
