
Losartan对高氧致慢性肺疾病新生大鼠肺纤维化的干预作用
2010-08-21 13:46陈宁张丹单丽沈刘雪雁尚云晓薛辛东
中国医科大学学报 2010年8期
陈宁,张丹,单丽沈,刘雪雁,尚云晓,薛辛东
(中国医科大学 附属盛京医院小儿呼吸内科,沈阳 110001)
新生儿慢性肺疾病(chronic lung disease,CLD)作为早产儿肺透明膜病治疗的并发症在1967年首次被Northway描述,它也被称为支气管肺发育不良,以肺泡发育受阻、肺间质广泛纤维化为主要病理特征。肺纤维化的发生是由许多细胞因子、炎性介质、内皮细胞和上皮细胞等构成的复杂的网络。有研究表明,肺组织局部的肾素-血管紧张素系统可能是纤维化的发生中心环节,血管紧张素Ⅱ(angiotensinⅡ,AngⅡ)通过与血管紧张素Ⅱ1型受体(angiotensin Ⅱ type 1 receptor,AT1R)结合,调节细胞因子、细胞外基质沉积[1,2]并诱导肺泡上皮细胞凋亡[3],从而破坏正常的肺泡结构和功能,促进肺纤维化的发生。AT1R拮抗剂losartan可减轻博莱霉素诱导的纤维化动物模型的肺组织胶原的沉积[4,5]。近年研究表明,肾素-血管紧张素系统可能参与CLD 的发病过程[6,7],特异性 AT1R 拮抗剂 losartan可明显降低高氧致CLD新生大鼠肺组织羟脯氨酸的表达[8]。本研究进一步观察AT1R拮抗剂losartan对高氧致新生大鼠CLD肺组织胶原表达的影响,从而探讨高氧致CLD可能的发病机制。
1 材料与方法
1.1 材料
动物模型的制备:将Wistar新生大鼠160只(中国医科大学附属盛京医院实验动物中心提供)出生后(连同母鼠)24 h内随机分为4组:空气组(组Ⅰ)、高氧组(组Ⅱ)、高氧+注射用水组(组Ⅲ)和高氧+losartan组(组Ⅳ)。组Ⅰ放置于空气环境(FiO2为21%)中饲养。组Ⅱ~组Ⅳ置于氧箱中,持续输入氧气,维持氧浓度85%~90%,每日用测氧仪监测(FiCO2<0.5%,钠石灰吸收CO2)。每日定时开箱0.5 h称重、添水和饲料及更换垫料,并与组Ⅰ交换母鼠以免因氧中毒而导致代母鼠喂养能力下降。各组饲养条件均为室温22~25℃,湿度60%~70%。在生后第6日开始,组Ⅳ每日将losartan(5 mg/kg)加到注射用水中按3 ml/kg灌胃,组Ⅲ每日给等量注射用水灌胃,至实验结束。
1.2 方法
各组在实验后的1、3、7、14和21 d腹腔内注射5%水合氯醛(6 ml/kg)麻醉后处死。立即打开胸腔,无菌条件下分离肺组织,取右肺各叶用冷生理盐水洗净残血,吸干水分,置于无RNase的Eppendorf管中于-80℃冰箱中保存;取左肺置于4%多聚甲醛固定,常规脱水、石蜡包埋,制成蜡块,待测。
1.2.1 HE染色光镜观察:每组动物各时间点随机抽取8张切片,每张切片光镜下(×200)随机选取5个视野观察病理改变。
1.2.2 Masson三联染色光镜观察:光镜下观察胶原纤维呈蓝色,胞浆呈红色,胞核呈黑蓝色。光镜下观察对比蓝色胶原纤维所占比例和分布情况。
1.2.3 逆转录聚合酶链反应(reverse transcription polymerase chain reaction,RT-PCR)半定量方法检测肺组织Ⅰ型胶原(collagen typeⅠ,ColⅠ)mRNA的表达:按说明书操作提取总RNA。半定量分析ColⅠmRNA的含量。扩增条件:94℃3 min,94℃40 s,52 ℃ 1 min,72 ℃ 90 s,35个循环后 72 ℃ 7 min;扩增产物201 bp。引物序列由上海博亚生物技术有限公司合成,上游:5′-TTCAGTGGCTCGCTTCGG-3′;下游:5′-TCCAGCACCAATCCCTTC-3′。
1.2.4 ELISA方法测定肺组织ColⅠ蛋白的含量:每组动物各时间点随机抽取6份标本,称50 mg湿重肺组织剪碎,冰盒内超声粉碎、制成10%组织匀浆,按说明操作,结果以每mg肺组织所含ColⅠ的含量表示(ng/mg)。ELISA试剂盒购自上海森雄科技有限公司,考马斯亮兰蛋白测定试剂盒购自南京建成生物工程研究所。
1.3 统计学分析
数据以±s表示。应用SSPS11.5统计软件进行统计学处理,多组间比较采用F检验,两样本均数间比较采用t检验。P<0.05为差异有统计学意义。
2 结果
2.1 光镜观察肺组织形态学改变
组Ⅰ7 d肺泡形态较规则,大小均匀,肺泡间隔稍厚;14和21 d肺组织进一步肺泡化,肺泡间隔变薄、肺泡隔增多、肺泡数量增多,21 d更为明显。组Ⅱ7 d肺泡隔及肺泡腔内见巨噬细胞、中性粒细胞等炎性细胞浸润,肺泡内出血,偶见肺不张,肺泡间隔增厚不明显,终末气腔扩张;14 d肺泡间隔较同期组Ⅰ增宽,肺间质细胞增多,肺泡数目减少,终末气腔扩张明显,偶见小灶肺实变;到21 d肺组织正常形态破坏,肺泡隔显著增厚,肺泡数目明显减少、结构简单,终末气腔扩张更为明显、部分肺泡萎陷,并且肺出血及肺实变多见。组Ⅲ与组Ⅱ改变相似。组Ⅳ在7 d与组Ⅱ区别不明显,14和21 d肺组织肺泡隔变薄,纤维化明显减轻,但肺泡腔没有明显缩小。见图1。
2.2 Masson染色观察胶原沉积的变化
蓝色的胶原纤维主要在支气管及血管壁的外周,肺泡壁有少许。组Ⅱ随高氧时间的延长,胶原纤维在支气管及血管壁的外周沉积增加;肺泡间质沉积在7 d后逐渐增加,在肺泡周围呈条索样增生,14和21 d同组Ⅰ比较蓝色胶原纤维所占比例明显增加。组Ⅳ蓝色胶原纤维所占比例较组Ⅱ减少且排列比较疏松,但仍明显高于组Ⅰ。见图2。
2.3 肺组织ColⅠ表达的动态变化
2.3.1 各组肺组织ColⅠ蛋白表达的变化:组Ⅱ、组Ⅲ和组ⅣColⅠ的含量均随日龄增加逐渐增加(P<0.01)。7 d时组Ⅱ、组Ⅲ和组Ⅳ肺组织中ColⅠ蛋白的含量略高于组Ⅰ(P>0.05),14和21 d时较组Ⅰ显著上升(P<0.01)。组Ⅳ在14 d时较组Ⅱ略有下降(P>0.05),21d时则显著下降(P<0.01)。见表1。

表1 各组肺组织ColⅠ蛋白含量的变化(±s,ng/mg,n=6)Tab.1 Changes of ColⅠ protein in lung tissue of each group(±s,ng/mg,n=6)

表1 各组肺组织ColⅠ蛋白含量的变化(±s,ng/mg,n=6)Tab.1 Changes of ColⅠ protein in lung tissue of each group(±s,ng/mg,n=6)
1)P<0.01 vs groupⅠ;2)P< 0.01 vs groupⅡ.
Group 1 d after birth 3 d after birth 7 d after birth 14 d after birth 21 d after birthⅠ 473.78±27.78 455.67±49.88 438.44±51.27 430.59±36.20 431.80±28.11Ⅱ 457.73±37.30 468.99±29.21 486.69±30.47 521.99±37.141) 746.99±74.831)Ⅲ 457.73±37.30 468.99±29.21 471.09±49.91 534.00±43.041) 720.46±64.931)Ⅳ 457.73±37.30 468.99±29.21 483.89±48.23 502.19±43.411) 600.69±49.441),2)
2.3.2 各组肺组织ColⅠmRNA表达的变化:RTPCR结果显示,组Ⅰ各时间点ColⅠmRNA表达无明显差异(P>0.05),组Ⅱ和组ⅢColⅠmRNA表达随日龄的增加显著上升。7、14和21 d时组Ⅱ、组Ⅲ和组Ⅳ较组Ⅰ明显上升(P<0.05或0.01)。组Ⅳ在14 d时较组Ⅱ略有下降,但差异不明显,21 d时则显著下降(P<0.01)。见表2,图3。
表2 各组肺组织ColⅠmRNA表达相对量的比较(±s,n=8)Tab.2 Comparison with the expression of ColⅠ mRNA in lung tissue of each group(±s,n=8)
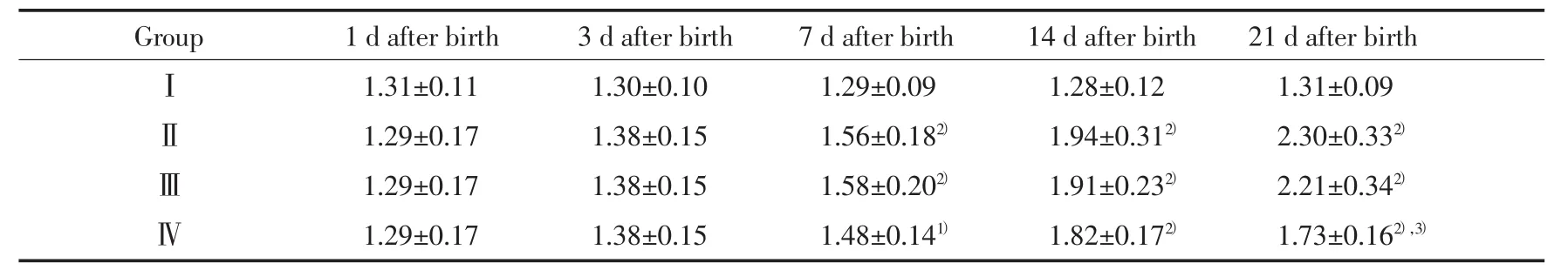
表2 各组肺组织ColⅠmRNA表达相对量的比较(±s,n=8)Tab.2 Comparison with the expression of ColⅠ mRNA in lung tissue of each group(±s,n=8)
1)P < 0.05,2)P < 0.01 vs groupⅠ;3)P < 0.01 vs groupⅡ.
Group 1 d after birth 3 d after birth 7 d after birth 14 d after birth 21 d after birthⅠ1.31±0.11 1.30±0.10 1.29±0.09 1.28±0.12 1.31±0.09Ⅱ1.29±0.17 1.38±0.15 1.56±0.182) 1.94±0.312) 2.30±0.332)Ⅲ1.29±0.17 1.38±0.15 1.58±0.202) 1.91±0.232) 2.21±0.342)Ⅳ1.29±0.17 1.38±0.15 1.48±0.141) 1.82±0.172) 1.73±0.162),3)
3 讨论
肺内细胞外基质成分主要包括胶原蛋白、弹性蛋白、蛋白聚糖及糖蛋白4种。细胞与细胞外基质蛋白之间复杂相互作用对于肺的细胞生长、分支发育及肺的成熟具有重要作用。胶原蛋白是肺组织主要的细胞外基质,约占肺脏干重的五分之一。肺内主要有6种胶原类型,其中ColⅠ和ColⅢ分布于肺间质,是主要的结构蛋白。正常肺组织发育过程中,适量的胶原的合成有助于组织细胞的分化成熟、毛细血管的正常排列,对于肺结构和功能的建立有重要的作用。在病理情况下胶原也参与创伤修复和器官纤维化的形成过程,是肺损伤后修复、呼吸道结构重建所必需的。

过去曾认为肺间质纤维化的发生是由于病变肺组织存在持续的肺泡炎,集中在炎性因子的研究,而没有考虑纤维化发生的初始因素。近年来研究认为上皮细胞损伤,上皮下成纤维细胞激活表型转化为成肌纤维细胞,导致细胞外基质异常沉积是肺纤维化的原因所在。而这些微环境损伤修复的细胞事件伴随着分子事件,即AngⅡ的产生。已经发现,在成人特发性纤维化、成人急性呼吸窘迫综合征、硅肺等纤维化肺疾病中以及纤维化动物模型中,肺组织中AngⅡ和血管紧张素转化酶浓度增加[9];并且AngⅡ以剂量依赖的方式促进成纤维细胞增殖。AngⅡ与成纤维细胞表面的AT1R结合,激活G蛋白,通过蛋白激酶C的级联放大激活丝裂原激活蛋白激酶,促进成纤维细胞增殖和分化,同时诱导成纤维细胞自分泌转化生长因子β[10]。应用AT1R拮抗剂losartan能够显著抑制AngⅡ诱导成纤维细胞的增殖与分化,抑制纤维化肺组织转化生长因子β表达及胶原沉积[1]、延缓博莱霉素诱导大鼠肺纤维化的进程[11]。这些提示AngⅡ在组织损伤、纤维化过程中可能扮演较为重要的角色。然而Keogh[12]同样在博莱霉素诱导肺纤维化实验中却得出相反的结论,认为肺纤维化疾病的发生是不依赖AngⅡ的诱导发生的,肺纤维化不能依靠losartan作为治疗策略。Chen[13]在研究百草枯诱导的肺纤维化时发现,纤维化肺组织中转化生长因子β及胶原含量增加与肺局部肾素-血管紧张系统无关。造成分歧的原因可能与不同品系的动物遗传背景不同、纤维化发生的诱因不同以及药物的剂量等因素所致。
近来研究发现在高氧CLD发病过程中也伴有肺局部肾素-血管紧张系统的激活与参与。已报道在高氧致新生大鼠CLD肺组织匀浆中AngⅡ、血管紧张素转化酶水平明显上升[6];并且AT1R在高氧致CLD肺组织表达亦显著增加[14]。本研究试图探讨AT1R拮抗剂losartan是否能阻止CLD的发生及其对高氧致CLD新生大鼠肺组织胶原沉积的影响。本研究的病理结果显示,在高氧后期肺泡间隔明显增宽、胶原沉积增加,losartan治疗后肺泡间隔变薄、胶原纤维排列比较疏松;但肺泡直径没有明显缩小,肺泡仍处于扩张状态,次级隔同组Ⅰ相比仍明显减少,呼吸膜面积并没有明显增加。进一步定量研究发现特异性AT1R拮抗剂losartan能够抑制高氧致CLD新生大鼠肺组织ColⅠ蛋白和mRNA的表达,由此可以推测losartan虽可以抑制高氧CLD新生大鼠肺组织胶原的沉积,但是并不能逆转高氧诱导的新生大鼠肺发育阻滞。因此,losartan并不能阻止高氧诱导CLD的发生。
[1]Marshall RP,Gohlke P,Chambers RC,et al.Angiotensin II and the fibroproliferative response to acute lung injury[J].Am J Physiol Lung Cell Mol Physiol,2004,286(1):L156-L164.
[2]Okada M,Suzuki K,Matsumoto M,et al.Effects of angiotensin on the expression of fibrosis-associated cytokines,growth factors,and matrix proteinsinhumanlungfibroblasts[J].JClinPharmTher,2009,34(3):288-299.
[3]Li X,Rayford H,Uhal BD.Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice[J].Am J Pathol,2003,163(6):2523-2530.
[4]Yao HW,Zhu JP,Zhao MH,et al.Losartan attenuates bleomycin-i nduced pulmonary fibrosis in rats[J].Respiration,2006,73(2):236-242.
[5]Molina-Molina M,Serrano-Mollar A,Bulbena O,et al.Losartan attenuates bleomycin induced lung fibrosis by increasing prostaglandin E2 synthesis[J].Thorax,2006,61(7):604-610.
[6]李玖军,李洪鹏,薛辛东.高氧致慢性肺疾病新生大鼠肺组织肾素–血管紧张素系统和TGF-β1的动态变化及卡托普利的干预机制[J].中国医科大学学报,2009,38(2):83-86.
[7]李玖军,陈宁,薛辛东.高氧致CLD新生大鼠肺组织肾素–血管紧张素系统的动态变化及其意义[J].中国现代医学杂志,2006,16(16):2423-2425.
[8]陈宁,李玖军,薛辛东.洛沙坦对高氧致慢性肺疾病新生大鼠肺纤维化的影响[J].中国当代儿科杂志,2007,9(6):591-594.
[9]Marshall RP,Mc Anulty RJ,Laurent GJ.Angiotensin Ⅱ is mitogenic for human lung fibroblasts via activation of the type 1 receptor[J].Am J Respir Crit Care Med,2000,161(6):1999-2004.
[10]Wenzel S,Taimor G,Piper HM,et al.Redox-sensitive intermediates mediate angiotensin Ⅱ-induced p38 MAP kinase activation,AP-1 binding activity,and TGF-beta expression in adult ventricular cardiomyocytes[J].J FASEB,2001,15(12):2291-2293.
[11]Mancini GB,Khalil N.Angiotensin Ⅱ type 1 receptor blocker inhibitspulmonaryinjury[J].ClinInvestMed,2005,28(3):118-126.
[12]Keogh KA,Standing J,Kane GC,et al.Angiotensin Ⅱ antagonism fails to ameliorate bleomycin-induced pulmonary fibrosis in mice[J].Eur Respir J,2005,25(4):708-714.
[13]Chen CM,Chou HC,Hsu HH,et al.Transforming growth factorbeta1 upregulation is independent of angiotensin in paraquat-induced lung fibrosis[J].Toxicology,2005,216(2-3):181-187.
[14]陈宁,刘雪雁,富建华,等.高氧致新生大鼠慢性肺损伤肺组织血管紧张素Ⅱ1型受体表达的变化[J].中国医科大学学报,2008,37(2):162-164.
猜你喜欢
中老年保健(2022年2期)2022-11-25
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年4期)2021-07-23
东方考古(2021年0期)2021-07-22
摄影之友(影像视觉)(2020年2期)2021-01-14
渔业研究(2018年5期)2018-01-17
中国卫生标准管理(2015年15期)2016-01-15
中国继续医学教育(2015年3期)2016-01-06
医学美学美容·中旬刊(2015年2期)2015-10-21
中国医疗美容(2015年4期)2015-04-27
