
把分子模拟法引入高分子物理实验教学
2010-07-02 00:34:43朱平平何平笙杨海洋梁好均
大学化学 2010年4期
朱平平何平笙杨海洋梁好均
(1中国科学技术大学实验教学中心 安徽合肥230026;2中国科学技术大学高分子科学与工程系 安徽合肥230026)
计算机与化学
把分子模拟法引入高分子物理实验教学
朱平平1,2何平笙2杨海洋1,2梁好均2
(1中国科学技术大学实验教学中心 安徽合肥230026;2中国科学技术大学高分子科学与工程系 安徽合肥230026)
分子模拟法(也被称作计算机模拟法)是用计算机以原子水平的分子模型来模拟分子的结构与行为,进而模拟分子体系的各种物理与化学性质[1],它已成为一种重要的科学研究方法,并且被广泛地应用于高分子科学的研究中[2-3]。作者在国内首先提出把计算机技术用于高分子物理实验教学[4-8],并先后开发出5个高分子物理的计算机模拟实验,是高分子物理实验教学的创新模式,即在实验教学中采用分子模拟法模拟和研究现代物理实验方法不能直接研究或观测的高分子物理现象及过程,取得了很好的教学效果。下面对这5个实验进行简单介绍。
1 用分子模拟(MP)软件构建全同立构聚丙烯分子、聚乙烯分子并计算它们末端的直线距离
小分子化合物不存在形态的问题;高分子链的构象、形态是高分子特有的问题,也是教学的重点和难点之一。由于单键的内旋转,使得柔性大分子在某一瞬间的构象与另一瞬间不同,链构象数很大,链的形态不断改变,尺寸也随之发生变化。然而高分子形态的研究主要是间接利用高分子稀溶液性质,因此通常的高分子物理实验难以直接涉及高分子链的构象及形态等问题。这个实验的开设可使学生对高分子链构象形态问题有直观、形象、透彻的了解,能弥补实验教学的不足。
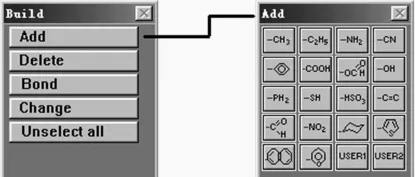
图1 [Build]菜单和其中的可加成的分子片段
用分子模拟(MP)软件可以在计算机屏幕上直接构建(合成)高分子长链,如构建聚丙烯分子,从主菜单窗口中选择[Build],出现构造[Build]菜单窗口,再选择[Add]便出现有各分子基团的窗口(图1),从中选取乙基片段,用鼠标器标亮其中的一个氢原子,从[Add]菜单窗口中选取甲基片段,至此完成了丙烷分子的构建。重复以下的操作:用鼠标器标亮其中的一个氢原子,从[Add]菜单中选择甲基和乙基片段,即可完成聚丙烯分子的构建。这里重要的是要选对合适的氢原子,否则得到的就不是全同立构聚丙烯分子,而是无规立构聚丙烯分子。另外,还要从主菜单窗口中选择[Build],再选择[Change],用鼠标器标亮扭转角的4个原子,将Torsion角调整为180°、60°、180°、60°……即TGTG……的构象,即可得到全同立构聚丙烯分子的螺旋形构象,如图2所示。

图2 全同立构聚丙烯分子的螺旋形构象
相同的方法还可以构建聚乙烯分子。很容易想象,如果所有相邻的3个C—C单键都取反式的构象,则高分子链是呈锯齿形的伸直链。图3是用分子模拟软件构建的主链含100个碳原子的聚乙烯伸展链,右边的局部放大图清楚地显示主链呈锯齿形。但当有些相邻的3个单键取左、右式构象时,高分子链则发生了弯曲,从主菜单窗口中选择[Build],再选择[Change],用鼠标器标亮扭转角的4个原子,调整Torsion角,实际就是通过内旋转改变链的构象,即可得到弯曲的聚乙烯链,如图4显示的是弯曲程度不同的两条聚乙烯链,也可以认为是同一条柔性链在不同时刻的形态。
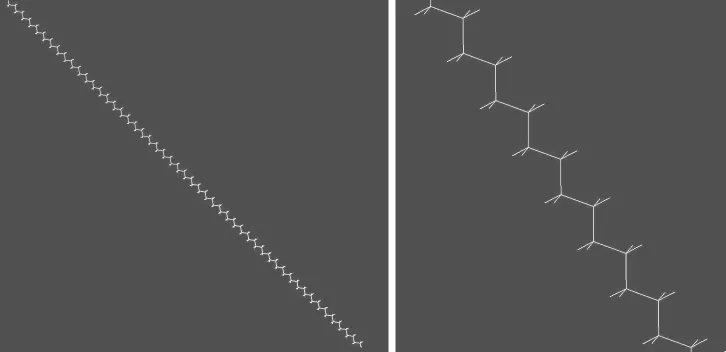
图3 主链含100个碳原子的聚乙烯伸展链
标亮第一和最后一个碳原子,选择[Analyse],再选择[Measure],这时得到的数据即是该聚乙烯分子链的末端距。通过多次内旋转,再测量它的末端距,从中来理解内旋转对高分子链末端距的极大影响。既有屏幕上的直观形象,又有真实测量值。
2 用分子模拟(MP)软件计算聚丙烯酸甲酯的构象能量
前一个实验可使学生对高分子链构象、形态及尺寸问题有直观、形象的认识。实际上,高分子链的内旋转不是自由的,而是受到相邻链节中非键合原子或基团间相互作用的影响,内旋转是受阻的,构象能E(φ)与内旋转角φ之间有很大关系,是一个很复杂的函数。然而对于柔性高分子,即使不能自由内旋转,可以实现的构象数目仍是非常大的。对于一个主链上含有n个单键的柔性大分子,其构象将由(n-2)个内旋转角决定。

图4 主链含100个碳原子的聚乙烯链
依实验学时安排和计算机的运行速度,我们在教学中建议学生分别构建含3个和5个单体单元的聚丙烯酸甲酯片段,先后选择两个内旋转角,并按5°或10°的间隔来计算。如果构建的丙烯酸甲酯片段太长、或内旋转角数目多、或角度间隔小,会因所包含的数据太多而使计算机工作量过大,计算速度太慢。
选定4个原子,选按[Torsion one]之后,再用鼠标器选中第2个扭转角的4个原子,再按[Torsion two]和[Torsion RUN],在屏幕上可出现相应的对话框。输入相应的参数后,按[OK]钮,计算机即开始运行。由于是同时旋转两个二面角,计算的量也是很大的。若依10°间隔角度计,在-180°、-170°....170°、180°每一个角度都要进行另一个每隔10°间隔的计算。同样,当程序完成计算构象能之后,调用计算机已自动存档的Conforme.out文件可查看计算结果。文件中有3列数据,分别对应内旋转角1(φ1)、内旋转角2(φ2)和构象能(E),学生应按要求绘制E(φ)-φ图。
3 二维高分子链形态的计算机模拟
碳链高分子在内旋转时,相邻的3个C—C单键呈反式、左旁式和右旁式3种构象时位能最低,构象最稳定,这3种构象的分子又称为内旋转异构体。依此推算,对于一根主链含有10000个C—C单键的碳链高分子,可能的异构体数目为39998(≈104770),这是个非常大的数字。在高分子链的构象发生变化时,链的尺寸也随之变化,因此对高分子链的构象、形态、尺寸都需要作统计的描述。在数学处理上,常采用向量运算求取尺寸的平均值,如均方末端距(末端距平分的平均值)、均方回转半径以及它们的平方根(根均方末端距、根均方回转半径)。
决定形态的重要因素是高分子链的化学结构和链单元间的相互作用,在溶液中的高分子链形态还受溶剂和温度的影响。不同条件下高分子链的形态差别较大,需用不同的模型来描述,如:无规行走(简称RW)和自回避行走(简称SAW)。
作者应用自编的改进型四位置模型模拟二维空间中的SAW、RW链。原四位置模型用于二维格子中,每个链单元的中心位于格子的中心,要用4个格点才能表示这个链单元,比较复杂,算法程序编写也很繁琐。对四位置模型改进后,则是以格点作为链单元的中心,一根链单元只对应一个格点,即4个相邻格子的共同格点,每个链单元的位置用一个格点表示,简化了算法程序的编写。改进后的模型仍具有原模型的优点,即,在对SAW链进行抽样时只需检验体积排除条件和键长条件是否满足,如果都满足,键就不可能相交,也就没有必要检验键是否相交。
如设置一定链长,计算数目为100万次,采样间隔为1000。点击“开始”按钮开始计算,在程序主窗口中可直接观察到链的形态和尺寸在不断变化,在计算完成后,即可获得一定数量的不同形态的SAW链或RW链。图5是链长为50的自回避行走链的某个形态,“获得链数”中显示为373,因采样间隔为1000,表明共生成了约373000个链,但是只显示其中的373个链,若在“输入”框中输入20,显示的就是这373个链中的第20个。界面右下部显示的是均方末端距和均方回转半径。
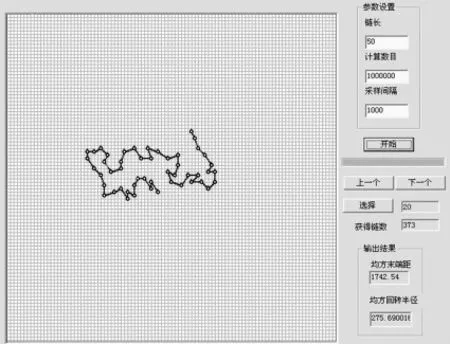
图5 自回避行走链
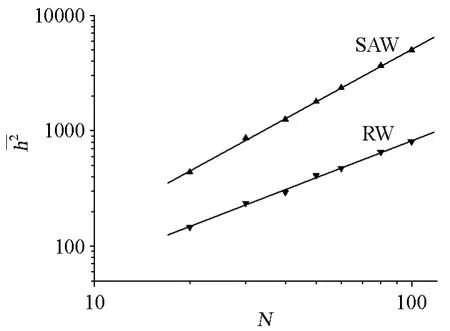
图6 均方末端距-聚合度图
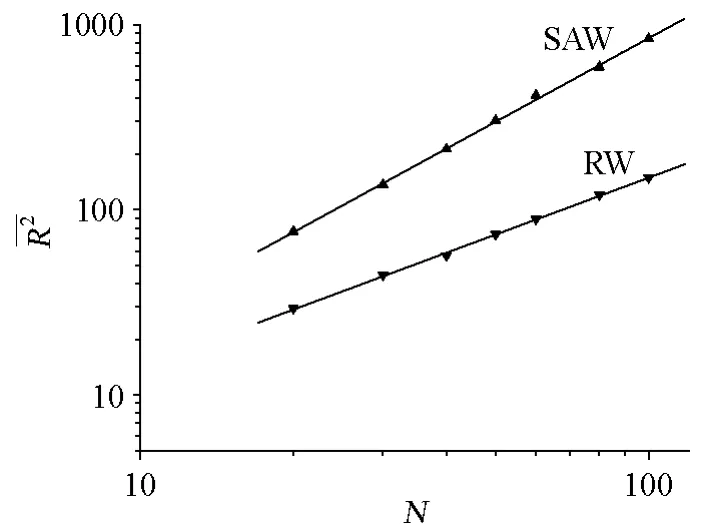
图7 均方回转半径-聚合度图
4 受限空间中的高分子链穿越纳米管道的M onte Carlo模拟
利用Monte Carlo方法模拟受限空间中的高分子链如何在熵驱动下穿越纳米管道,涉及到高分子物理中高分子尺寸、构象熵等基本概念和知识点,对学生理解构象熵这一概念在高分子物理学习中的特殊重要性非常有帮助。
受限空间中高分子穿越纳米管道过程中,由于熵的作用,高分子自发地从熵受限的区域迁移到受限程度较低或完全不受限的区域[10]。如图8所示,当高分子从区域A进入纳米管道以后,高分子的构象熵会不断地减小,由此导致自由能升高。而当高分子完全穿越纳米管道后,高分子的构象熵又会进一步增加,导致自由能减小。由于区域A比区域B更加受限,因此高分子在区域B的自由能要低于在区域A的自由能。由于高分子在从区域A自发迁移到区域B时要越过一定的自由能壁垒,高分子穿越自由能壁垒对应的是能量升高的过程,因此高分子成功穿越的概率并不高。如果把高分子从开始穿越到最终又回到起点的时间定义为τtrap,则自由能壁垒ΔE越大,τtrap越长。如果把高分子穿越管道的过程与化学反应过程相类比,则τtrap与化学反应速率常数k之间满足关系:τtrap~k-1。根据阿仑尼乌斯(Arrhenius)公式,化学反应速率常数k与反应活化能ΔE之间满足关系:k~e-ΔE/KT。由此得到τtrap时间与自由能壁垒ΔE之间满足关系:lgτtrap~ΔE。构建不同长度的高分子链并穿越不同长度的纳米管道,通过测定τtrap,就可以讨论高分子穿越纳米管道的动力学过程。
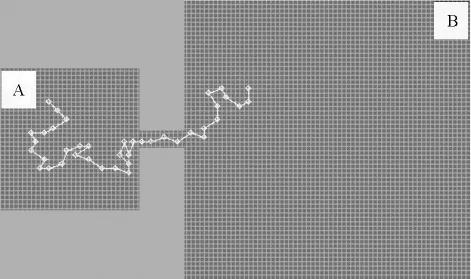
图8 受限空间中的高分子链穿越纳米管道示意图
模拟结果表明,高分子链穿越的动力学过程与化学反应动力学过程具有相似性,很有新意,学生也很感兴趣。此实验为我校特有。
5 使用M onte Carlo方法观察受限状态下嵌段聚合物自组装结构
大分子自组装是一种以高分子为组装单元的超分子行为,属于超分子化学和高分子科学的交叉学科,而其中嵌段共聚物的自组装则是大分子自组装领域的主流。为了让学生及时了解此领域的研究成果,作者开发了本实验,适用于对称二嵌段共聚物受限在平行板间的自组装模拟[11]。在没有受限的本体中,对称的二嵌段共聚物自组装形成无定向排列的层状结构。而在平行板间自组装时,受平行板板壁的影响,二嵌段共聚物形成的层状结构可以平行或垂直于平行板排列。图9是嵌段聚合物分子自组装模拟软件界面,两种颜色代表两种不同的分子链段。
这个实验的开设对学生理解高分子物理中的高分子凝聚态结构、高分子之间相互作用、微观相分离等知识点有直观的帮助。
计算机模拟无疑是事半功倍的,由于实验涉及的知识点都是高分子科学中最基本、最重要的问题,也是高分子物理课程学习的重点、难点问题(表1),因此可起到实体分子模型和课堂教学不可能达到的效果。
此外,计算机模拟实验具有如下特点:
(1)一旦计算机和软件具备,平时几乎不需要维持费用,对实验教学是很有利的;
(2)保证了实验教学内容的先进性和新颖性;
(3)具有普及性,非常适于推广。
作者通过提供电脑程序源代码、发表介绍实验内容的论文和组织召开研讨班作示范教学,有关的计算机模拟实验已被国内多所高校所采用。
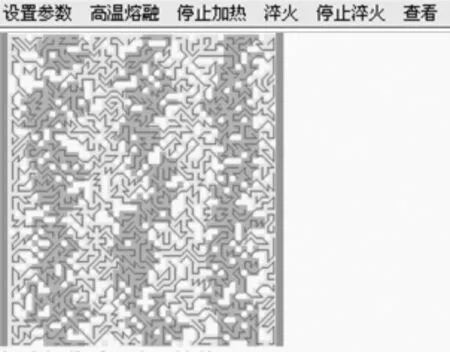
图9 高分子自组装模拟软件界面
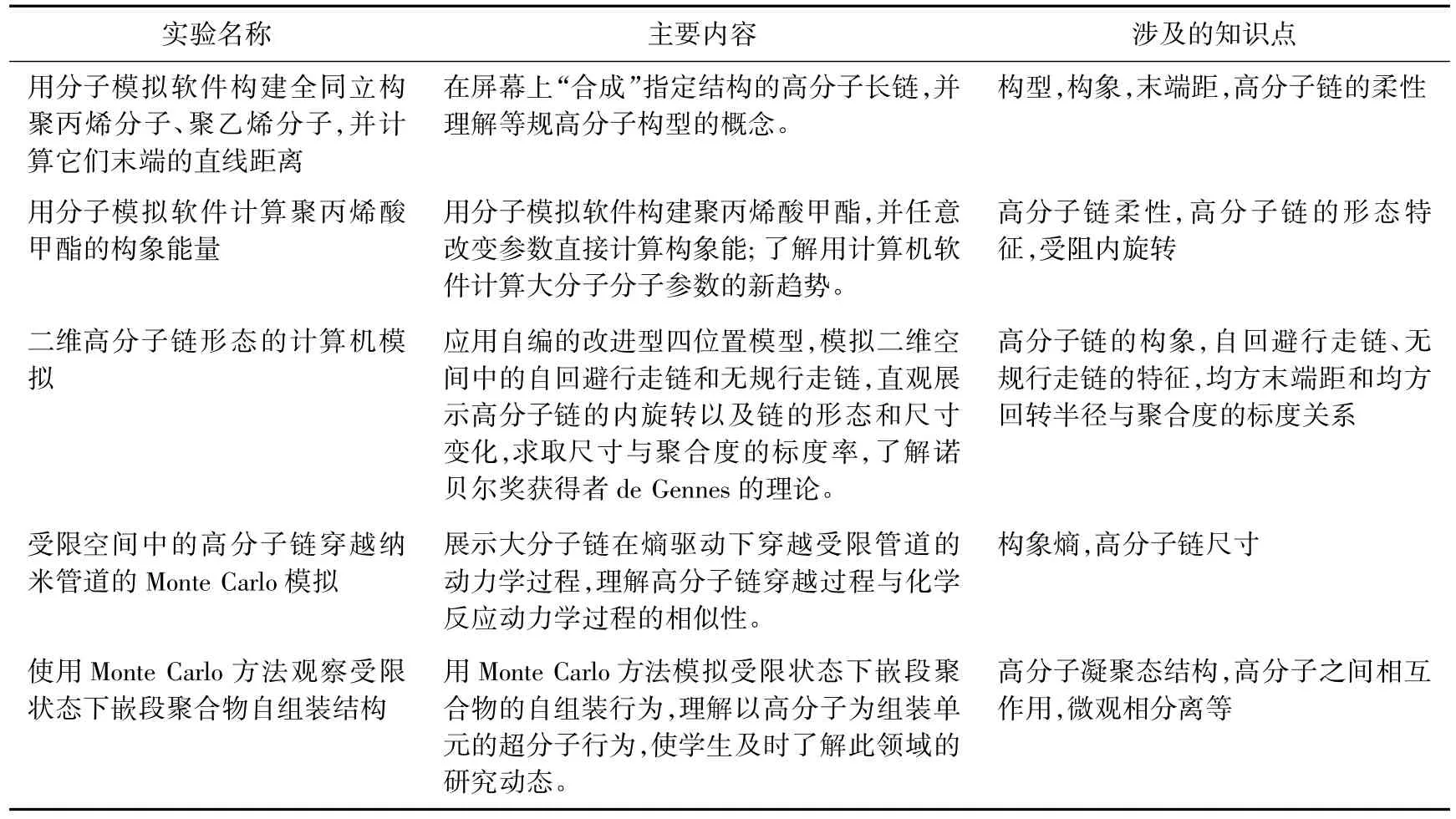
表1 开发的高分子物理计算机模拟实验
[1] 杨小震.分子模拟与高分子材料.北京:科学出版社,2002
[2] 雷军,张振军,刘斌,等.高分子通报,2005(6):122
[3] 杨玉良,张红东.高分子科学中的Monte Carlo方法.上海:复旦大学出版社,1993
[4] 何平笙,杨小震.高分子通报,2000(1):86
[5] 何平笙,李春娥.高分子通报,2000(2):94
[6] 杨海洋,易院平,朱平平,等.高分子通报,2003(5):76
[7] 杨海洋,朱平平,何平笙.高分子物理实验.第2版.合肥:中国科学技术大学出版社,2008
[8] 朱平平,杨海洋,何平笙.高分子通报,2007(7):61
[9] de Gennes P J.Scaling Concepts in Polymer Physics.Ithaca:Cornell University Press,1979
[10] Xie Y J,Yu H T,Yang H Y,et al,Biochemical and Biophysical Research Communications,2006,349:15
[11] Chen P,He X H,Liang H J,Journal of Chemical Physics,2006,124:104906
猜你喜欢
功能高分子学报(2022年5期)2022-10-19 06:29:44
功能高分子学报(2022年5期)2022-10-19 06:29:44
功能高分子学报(2022年4期)2022-08-05 03:05:40
功能高分子学报(2022年4期)2022-08-05 03:05:40
原子与分子物理学报(2021年2期)2021-03-29 07:31:16
纺织科技进展(2016年3期)2016-11-29 01:27:04
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
中国资源综合利用(2016年6期)2016-01-22 07:28:55
应用化工(2014年11期)2014-08-16 15:59:13
应用化工(2014年7期)2014-08-09 09:20:23
