
普乐安片氮含量测定的不确定度分析
2010-06-01 07:53:22黄燕萍
中国药业 2010年1期
黄燕萍
(广西壮族自治区北海食品药品检验所,广西 北海 536000)
笔者参考文献[1-4]对普乐安片的氮含量进行了测定,分析了测定过程的测量不确定度来源,并对各不确定度分量进行评定,提出了该测定方法的合成不确定度,报道如下。
1 仪器与试药
AW-120 型电子天平(Max=120 g,d=0.1 mg);FOSS TecatorTMDigestor消化炉;FOSS KjeltecTM2200型半自动定氮仪;50 mL及25 mL滴定管。普乐安片(市售品);碳酸钠纯度标准物质(编号为BS2307,定值日期为2008年3月,碱量纯度标准值为99.99%,扩展不确定度<k=2>为0.02%);硫酸铵(优级纯,天津市光复精细化工研究所);硫酸标准滴定溶液(0.05 mol/L);其他试剂为分析纯。
2 方法与结果
2.1 滴定液配制与标定[4]
取硫酸3 mL,缓缓注入适量水中,冷却至室温,加水稀释至1 000 mL,摇匀,即得硫酸滴定液(0.05 mol/L)。取 270 ~300 ℃干燥至恒重的碳酸钠纯度标准物质约0.15 g,精密称定,加水50 mL使溶解,加甲基红-溴甲酚绿混合指示液10滴,用硫酸滴定液滴定至溶液由绿色转变为紫红色时,煮沸2 min,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1 mL硫酸滴定液(0.05 mol/L)相当于5.30 mg的碳酸钠纯度标准物质。根据硫酸滴定液的消耗量与碳酸钠纯度标准物质的取用量,算出硫酸滴定液的浓度。
2.2 试样处理与测定
取本品10片,除去包衣,精密称定,研细,取约0.6 g,精密称定,移入干燥的FOSS消化瓶中,加入硫酸铜0.2 g、硫酸钾6 g、硫酸10 mL,摇匀,置消化炉中,于250℃低温消化1.5 h,再升温至400℃,继续消化2 h即消化完全,放冷备用。同法消化试剂,作为空白对照液。向接受瓶内加入10滴混合指示液(1份1 g/L的甲基红乙醇溶液与5份1 g/L的溴甲酚绿乙醇溶液,临用时混合),取消化液,置FOSS KjeltecTM2200型蒸馏仪上依法蒸馏[接受瓶加50 mL接受液(4%硼酸溶液),消化管加50 mL强碱液(40%氢氧化钠溶液)和80 mL水];同时进行试剂空白消化液的蒸馏处理。取接收瓶,以硫酸滴定液(0.05 mol/L)滴定至灰紫色为终点,记录消耗的硫酸滴定液体积;同时滴定试剂空白蒸馏液。
2.3 测量数学模型
浓度计算公式:称取样品0.6 g进行消化,消化完全后在半自动凯氏定氮仪上蒸馏,滴定,按硫酸滴定液消耗量计算氮的含量。在整个测定过程中,认为消化时消化完全和硼酸溶液吸收氨是完全的,故测量数学模型为公式1。

式中:N为试样含氮量(mg/片),m为取样量(g),v2为滴定试样时所需硫酸滴定液的体积(mL),v1为滴定空白对照液时所需硫酸滴定液的体积(mL),c为硫酸滴定液实际浓度(mol/L),0.05为硫酸滴定液的标示浓度(mol/L),1.401为氮的摩尔质量(mg/mol)。
不确定度传播律:采用的标准不确定度合成公式为公式2。
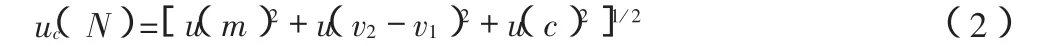
2.4 不确定度分量量化
2.4.1 u( c)
硫酸滴定液引入的不确定度主要有以下3种。
1)碳酸钠纯度标准物质引入的不确定度
实验室使用的碳酸钠纯度标准物质的碱量纯度标准值 c=99.99%,扩展不确定度为0.02%,k=2,则标准物质引入的不确定度u(m1)=0.02%/2=0.01%,相对不确定度urel(m1)=u(m1)/c=0.01%/99.99%=1×10-4。碳酸钠纯度标准物质的取样量为0.15g,天平使用说明书给出的非线性误差为±0.1mg,按均匀分布考虑,k=31/2,称重按照减量法,则 u(m2)=21/2×0.1/31/2=0.08165mg;称样量m=0.149 35 g,则相对不确定度urel(m2)=u(m2)/m=0.081 65/(0.149 35 ×103)=5.467 ×10-4。因此,碳酸钠纯度标准物质引入的合成不确定度为urel(m)=[urel(m1)2+urel(m2)2]1/2=[(1 × 10-4)2+(5.467 × 10-4)2]1/2=5.558 × 10-4。
2)50 mL滴定管引入的不确定度
经过校准的50 mL A级滴定管允差为±0.05 mL[5],按三角分布,取k=61/2,则50 mL滴定管示值引入的不确定度u1(v)=0.05/61/2=0.020 4,相对不确定度u1 rel(v)=0.020 4/50=4.08×10-4。因滴定管校准的温度为20℃,实际滴定温度为28℃,膨胀系数α=2.1×10-4,滴定管体积50 mL,按照矩形分布计算,故 k=31/2,u(2v)=50×8×2.1×10-4/31/2=0.048 5;消耗量(v)为27.98 mL,故相对不确定度u2r(elv)=0.048 5/27.98=0.001 733。读数时的标准不确定度约为0.02 mL,按照均匀分布计算,k=31/2,u3r(elv)=0.02/(50×31/2)=2.309 ×10-4。因此,滴定管引入的合成不确定度ur(elv)=[u1rel(v)2+u2rel(v)2+u3rel(v)2]1/2=([4.08×10-4)2+0.001 7332+ (2.309 × 10-4)2]1/2=1.795 × 10-3。
3)硫酸滴定液重复标定引入的不确定度。
硫酸滴定液6次重复标定结果分别为0.050 001,0.049 998,0.049 988,0.050 036,0.049 957,0.049 999 mol/L,平均值=0.049 997 mol/L。由贝塞尔公式计算得,测量结果以平均值来表示,故 (urep)=(srep)/n1/2=1.037×10-5,相对不确定度ur(elrep)=1.037×10-5/0.049997=2.074×10-4。因此,硫酸滴定液重复标定引入的不确定度ur(elc)=[urel(m)2+urel(v)2+urel(rep)2]1/2=([5.558×10-4)2+(1.795 × 10-3)2+ (2.074 × 10-4)2]1/2=1.890 × 10-3。
2.4.2 u( m)
主要考虑称量引入的不确定度。用天平称量样品0.6 g,天平使用说明书给出的非线性误差为±0.000 1 g,按均匀分布考虑,k=31/2,称重按照减量法,则 um=21/2×0.000 1/31/2=8.165 ×10-5g;称样量m=0.610 4 g,则相对不确定度ur(elm)=um/m=8.165×10-5/0.610 4=1.338×10-4。
2.4.3 u( v2-v1)
主要考虑25 mL滴定管引起的不确定度。经过校准的25 mL A级滴定管允差为±0.04 mL[5],按三角分布,取 k=61/2,则25 mL滴定管示值引入的不确定度 u1(v)=0.04/61/2=0.016 3,相对不确定度 u1rel(v)=0.016 3/25=6.520×10-4。因滴定管校准的温度为20℃,实际滴定温度为26℃,膨胀系数 α=2.1×10-4,滴定管体积25 mL,按照矩形分布计算,故 k=31/2,u2(v)=25×6×2.1×10-4/31/2=0.018 2;消耗量(v2-v1)为 13.41 mL,则 u2rel(v)=0.018 2/13.41=1.357×10-3。读数时的标准不确定度约为0.02 mL,按照均匀分布计算,k=31/2,u3rel(v)=0.02/(25 ×31/2)=4.619 ×10-4。硫酸标准滴定液消耗量取值为 v2-v1,则滴定管引入的合成不确定度urel(v2-v1)=21/2[u1r(elv)2+u2r(elv)2+u3r(elv)2]1/2=21/2[(6.520×10-4)2+ (1.357 × 10-3)2+ (4.619 × 10-4)2]1/2=2.227 × 10-3。
2.4.4 u(N)
滴定体积的重复性通过重现性试验考察。用凯氏定氮法对样品进行6次重复测定,结果样品氮含量分别为17.06,16.95,17.01,16.98,17.01,16.97 mg/片,为17.00 mg/片。由贝塞尔公式计算得 s(N)=0.039 0,测量结果以平均值来表示,故 u(N)=s(N)/n1/2=0.015 9,相对不确定度 urel(N)=0.015 9/17.00=9.353 ×10-4。
由于本测定方法是取供试品10片称定质量,计算求出其平均值,因此供试品平均片重的不确定度来源于10片重的称定。天平引入的不确定度=u1(m)=21/2×0.000 1/31/2=8.165×10-5g。样品平均片重称定 10次分别为 0.514 7,0.514 2,0.514 9,0.514 6,0.514 8,0.514 8,0.514 9,0.514 5,0.514 8,0.514 6 g,=0.514 7。按贝塞尔公式计算其标准不确定度,)=2.16×10-4g。因称量采用减重法,故其平均片重的相对标准不确定度
对5个样品进行加样回收试验,原样品中的平均氮含量为17.0 mg/片,RSD=0.32%,平均加标氮含量是17.003 m(g即称取0.080 18 g的硫酸铵),加标后样品中平均氮含量为34.0 mg/片,氮含量平均回收率为99.51%,RSD=0.15%。加入的标准物质浓度所引起的不确定度很小,可以忽略。每个样品都以相同的浓度添加,故可用加标后样品分析结果的标准不确定度与原始样品分析结果的标准不确定度合成平均回收率的标准不确定度
2.5 合成标准不确定度计算
测定各分量的不确定度相互独立,将上述数据合成,得样品含量的相对合成标准不确定度 ucrel=[urel(c)2+urel(m)2+urel(v2-v1)2+urel(N)2+urel)2+urel)2]1/2=[(1.890 ×10-3)2+ (1.338 ×10-4)2+(2.227 × 10-3)2+ (9.353 × 10-4)2+ (4.407 × 10-4)2+0.018 92]1/2=0.019 2,转化成绝对不确定度,u=c(N)·ucrel=0.019 2×17.0=0.326。
2.6 扩展不确定度计算
简易评定,取 k=2,则扩展不确定度 U=ku=2×0.326≈0.7。
2.7 测量结果表示
样品氮含量可表示为(17.0 ±0.7)mg/片,k=2。
3 讨论
本试验中滴定液的标定和样品的消化、蒸馏、滴定均是重要环节,应规范操作,使不确定度尽可能降低。从分析结果看,在影响检测结果的各个分量中,称量、平均片重和重复性对结果的影响很小,可以忽略不计;滴定液的浓度、量具等引入的不确定度相对显著,在分析了不确定度后,采取相应措施,误差可以进一步减小。通过对不确定度进行分析,可以为合理改进分析方法,将检测方案误差降至最小提供可靠依据。在进行合格的判定时,应考虑到不确定度,把不确定度加入判定结果,从而提高药品检验结果的准确性和可信度,保证结果的公平、公正。
[1]JJF1059-1999,测量不确定度评定与表示[S].
[2]中国合格评定国家认可委员会.化学分析中不确定度的评估指南[M].北京:中国计量出版社,2006:1-148.
[3]国家药典委员会.中华人民共和国卫生部药品标准·中药成方制剂(第十四册)[M].北京:人民卫生出版社,1997:180.
[4]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:附录 49,附录 103.
[5]JJG196-2006,国家计量检定规程·常用玻璃量器 [S].
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28 06:26:50
——以“NaOH标准溶液的标定”微课教学为例
卫生职业教育(2019年7期)2019-04-04 05:30:16
童话世界(2017年29期)2017-12-16 07:59:32
试题与研究·高考理综化学(2016年1期)2017-05-05 16:56:54
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
中小学实验与装备(2015年5期)2016-04-19 06:53:35
化学教学(2015年8期)2015-10-15 01:58:48
应用化工(2014年11期)2014-08-16 15:59:13
江苏大学学报(自然科学版)(2014年6期)2014-02-28 01:32:43
中国氯碱(2014年10期)2014-02-28 01:05:01
