
康儿灵颗粒质量标准研究
2010-02-07 03:48唐全红胡玉霞高柏丽
中成药 2010年9期
和 芳, 唐全红, 胡玉霞, 高柏丽
(北京创立科创医药技术开发有限公司,北京100029)
康儿灵颗粒是由刺五加、白术(炒)、莲子、六神曲(炒)、茯苓、麦芽(炒)、陈皮、枳壳(炒)、山楂(炒焦)、甘草(炙)、胡黄连、使君子十二味中药制成的中药复方颗粒,它的质量标准收载于《中华人民共和国卫生部药品标准》中药成方制剂第13册,标准编号:WS3-B-2613-97。功能主治为益气健脾,开胃消食,用于脾胃虚弱,食欲不振,消化不良,形体瘦弱[1]。此标准为卫生部药典委员会于1997年8月编辑出版,由于此标准的起草年代较早,技术水平和条件有限,所以该标准非常简单,控制项目很少,质量标准中仅对陈皮、枳壳中所含有的橙皮苷进行了薄层色谱鉴别,对其它药味没有任何控制。为了更有效地控制该药品的药品质量,我们参考文献[2-3]新建立了刺五加、白术、莲子、胡黄连的薄层色谱鉴别方法,并参考文献[2]新建立了陈皮和枳壳中的橙皮苷的高效液相含量测定方法,重新制订了质量标准,使其中的六味药材得到了有效地控制,使药品的质量更好地得到了保证。
1 仪器与试药
1.1 仪器 LC-2010A高效液相色谱仪、PDA-10 Avp检测器(日本Shimazu公司),SB5200超声波清洗器(上海Branson公司);Milli Q超纯水机(法国Millipore公司)。
1.2 药品与试剂 硅胶G薄层板;硅胶GF254薄层板(青岛海洋化工厂分厂);橙皮苷、肉桂酸、异嗪皮啶、刺五加对照药材、白术对照药材、莲子对照药材(购自中国药品生物制品检定所);康儿灵颗粒及阴性样品(自制);甲醇(色谱纯);水为超纯水;其余试剂均为分析纯。
2 方法与结果
2.1 定性鉴别
2.1.1 刺五加的鉴别 取康儿灵颗粒10 g,研细,加水25 mL振摇使溶解,过滤,过滤液加氯仿萃取3次,每次20 mL,合并氯仿萃取液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取不含刺五加药材的阴性样品,同法制成阴性对照溶液;另取刺五加对照药材0.5 g,加氯仿25 mL,超声处理30 min,过滤,滤液蒸干,残渣加甲醇1 mL使溶解,作为对照药材溶液。再取异嗪皮啶对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。照中国药典薄层色谱法试验,吸取上述4种溶液各10 μL,分别点于同一硅胶G薄层板上,以氯仿-石油醚(60~90℃)-甲醇(9.5∶0.2∶0.4)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。在供试品色谱中,与对照药材及对照品色谱相应的位置上,显相同颜色的荧光斑点。结果见图1。
2.1.2 白术的鉴别 取康儿灵颗粒15 g,加正己烷40 mL,超声处理15 min,滤过,滤液挥至0.5 mL,作为供试品溶液。另取白术对照药材5 g,同法制成对照药材溶液。照中国药典薄层色谱法试验,吸取上述新制备的供试品溶液15 μL,对照药材溶液10 μL,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(9∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,热风吹至斑点清晰。在供试品色谱中,与对照药材色谱相应的位置上,显相同颜色的斑点。结果见图2。
2.1.3 莲子的鉴别 取康儿灵颗粒15 g,加氯仿50 mL,超声处理30 min,放置过夜,滤过,滤液蒸干,残渣加乙酸乙酯1 mL使溶解,作为供试品溶液。另取莲子对照药材5 g,同法制成对照药材溶液。照中国药典薄层色谱法试验,吸取上述两种溶液各6 μL,分别点于同一硅胶G薄层板上,以正己烷-丙酮(7∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,置紫外光灯(365 nm)下检视。在供试品色谱中,与对照药材色谱相应的位置上,显相同颜色的荧光斑点。结果见图3。

图1 刺五加的薄层鉴别图

图2 白术的薄层鉴别图
2.1.4 胡黄连的鉴别 取康儿灵颗粒10 g,加乙醇40 mL加热回流2 h,滤过,滤液蒸干,残渣加1%氢氧化钠溶液20 mL溶解,转移至分液漏斗中,用乙醚振摇提取2次,每次20 mL,弃乙醚液,碱液用盐酸调节pH值至2~3,再用乙醚提取3次,每次15 mL,合并乙醚液,挥干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取肉桂酸对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。照中国药典薄层色谱法试验,吸取上述供试品溶液2 μL,对照品溶液1 μL,分别点于同一硅胶GF254薄层板上,以正己烷-乙醚-冰醋酸(5∶5∶0.1)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。在供试品色谱中,与对照品色谱相应的位置上,显相同颜色的斑点。结果见图4。

图3 莲子的薄层鉴别图

图4 胡黄连的薄层鉴别图
2.2 含量测定
2.2.1 色谱条件:色谱柱:Kromasil C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇 - 水 - 冰醋酸(35 ∶61 ∶4),流速:1.0 mL/min;检测波长:283nm;柱温:常温;理论板数按橙皮苷峰计算不低于3 000。
2.2.2 供试品溶液的制备:取康儿灵颗粒,研细,取2 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50 mL,称定重量,超声处理(功率250 W,频率20 kHz)30 min,取出,放冷,再称定重量,加80%甲醇补足减失的重量,摇匀,滤过,取续滤液用微孔滤膜(0.45 μm)滤过,即得。
2.2.3 专属性考察按处方中药味比例,自配不含陈皮和枳壳的群药,按其工艺制成阴性制剂,再按供试品溶液制备方法制备并测定,结果陈皮和枳壳阴性溶液在与橙皮苷对照品相同保留时间处未显色谱峰,表明阴性样品没有干扰。测定结果见图5。

图5 康儿灵颗粒HPLC色谱图
2.2.4 线性关系考察 精密量取橙皮苷对照品溶液(0.069 2 mg/mL)1、2、4、6、8、10 mL 分别置 10 mL 量瓶中,加甲醇至刻度,摇匀,分别精密吸取10 μL,按上述色谱条件测定,以峰面积为纵坐标,橙皮苷进样量(μg)为横坐标,绘制标准曲线。计算得回归方程为:Y=2 127 492X+1 817.7,r=0.999 9 结果表明橙皮苷在0.069 2 ~0.692 0 μg范围内线性良好。
2.2.5 精密度 精密吸取橙皮苷对照品溶液10 μL,重复进样6次,求得峰面积的相对标准偏差RSD为0.08%,表明仪器精密度良好。
2.2.6 稳定性考察 精密吸取同一供试品溶液(批号:20020301)10 μL,分别于配制后在 0、1、2、4、8、12、24 h,依法测定,记录峰面积,结果峰面积的RSD为1.68%。结果表明供试品溶液在24 h内基本稳定。
2.2.7 重复性试验 取康儿灵颗粒(批号为20020301)样品6份,按样品测定法测定,求得橙皮苷含量的 RSD为1.72%,说明本方法重复性良好。
2.2.8 回收率试验 采用加样回收法,称取已知含量的同一批的样品1 g,精密称定,分别精密加入橙皮苷对照品溶液(0.099 6 mg/mL)各5 mL,按供试品溶液的制备方法制备及上述色谱条件测定,计算回收率,结果见表1。
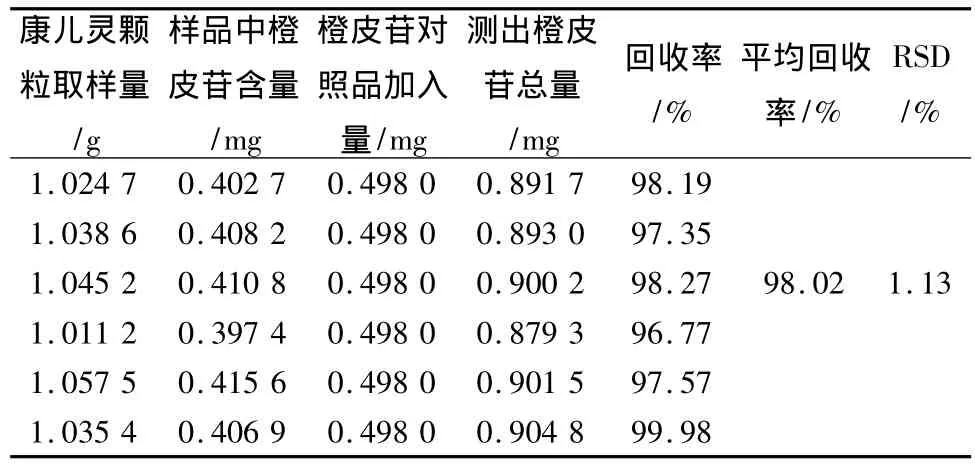
表1 加样回收率试验结果
2.2.9 样品含量测定 取各批样品分别按供试品溶液制备方法制备,按上述色谱条件测定峰面积,以外标法计算样品中橙皮苷的含量,结果见表2。

表2 样品测定结果
3 讨论
3.1 康儿灵颗粒处方中含有十二味药,原标准中已采用TLC法对陈皮和枳壳进行鉴别,现又增加了刺五加、白术、莲子、胡黄连四味药的TLC法定性鉴别;并且采用HPLC法对橙皮苷进行了含量测定。方法可靠,准确性好。可很好的控制药品质量。
3.2 曾摸索了方中麦芽的薄层色谱鉴别。康儿灵颗粒、麦芽对照药材取样量分别增大到20 g、10 g,按相应方法提取,点样,展开,检视。在供试品色谱中,与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性样品无干扰。但考虑取样量太大,故未收入康儿灵颗粒质量标准。
3.3 检测波长的选择:取橙皮苷对照品溶液,照色谱条件依法进样,对色谱峰在200~400 nm范围内测定光谱图,结果橙皮苷在283 nm处有最大吸收,故选择283 nm作为测定波长。
3.4 提取条件的选择 分别采用超声波提取(功率250 W,频率20 kHz)、热回流提取、冷浸提取,测定上述康儿灵颗粒中橙皮苷的含量,结果表明3种提取方法含量基本相近,基于超声波提取法方便快捷,故正文选择超声波提取法。
3.5 提取溶剂的选择 橙皮苷在甲醇中溶解度较好,分别以甲醇、80%甲醇、50%甲醇为溶剂,按供试品溶液制备方法分别制成供试品溶液,依法测定橙皮苷的含量,结果表明80%甲醇作为提取溶剂较好。
[1]中华人民共和国卫生部药品标准中药成方制剂第13册WS3-B-2613-97[S].1997.
[2]中国药典[S].一部.2005.
[3]卫生部药典委员会编著.中华人民共和国药典中药薄层色谱彩色图集[M].广东:广东科技出版社,1993.
猜你喜欢
今日农业(2022年2期)2022-11-16
今日农业(2021年6期)2021-06-09
特种经济动植物(2021年4期)2021-04-19
幸福·健康版(2018年3期)2018-03-23
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
海峡科技与产业(2016年3期)2016-05-17
云南中医学院学报(2015年3期)2015-07-31
中国当代医药(2015年24期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国当代医药(2015年8期)2015-03-01
