葛根芩连汤数字化色谱指纹谱的建立及其与组方药味的相关性研究
2010-01-30 06:23姚亚敏王新宏
中成药 2010年10期
安 叡, 姚亚敏, 王新宏
(1.上海中医药大学中药学院,上海201203;2.上海市公共卫生临床中心复旦大学附属公共卫生临床中心,上海201508)
葛根芩连汤为东汉医圣张仲景名方,出自《伤寒论·太阳篇》,由葛根、黄芩、黄连、炙甘草四味中药组成,有解表清里,升阳止泻功效,用于泻热下利[1]。指纹图谱作为控制中药复方质量的重要而有效的手段,与中药复方有效成分的复杂性、不确知性,以及发挥作用时众多成分的协同性、多靶点性特点相吻合[2]。但仅通过色谱方法得到的中药样品色谱图应该仅为样品的化学轮廓图,必须通过一定的数据处理和多个相关样品比较,指纹峰的归属鉴别等后才可以赋予色谱图以特征性。本研究采用梯度洗脱技术进行HPLC分离分析,建立葛根芩连汤的数字化色谱指纹谱,并分别对方中葛根、黄芩、黄连进行色谱指纹谱鉴别,探求各药材所呈现的特征峰群,建立复方与药材化学组成之间的联系,有助于提高中药复方质量控制水平。希望为阐明葛根芩连汤药效物质基础,从化学成分分析其配伍规律奠定基础。
1 仪器与试药
Agilent 1100型高效液相色谱仪(G1379A Degasser;G1311A Quantum;G1316A Column;G1314A Vwd);SK3200H超声仪(上海科导超声仪器有限公司)。
葛根、黄芩、黄连和炙甘草药材均购于上海康桥中药饮片有限公司,经本校赵志礼教授鉴定。
葛根素、大豆苷、大豆苷元、黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、盐酸巴马汀对照品(批号分别为110752-200511、111738-200501、111502-200101、 110715-200815、111595-200604、 111514-200403、 110713-200609、 110732-200506)均购自中国药品生物制品检定所;汉黄芩苷,批号061013;购自上海友思生物技术有限公司。
甲醇(HPLC级购于国药集团化学试剂有限公司),甲酸、三乙胺等试剂为分析纯,水为重蒸水。
2 方法与结果
2.1 色谱条件 色谱柱:Agilent TC-C18(4.6 mm×250 mm,5 μm);流动相:甲醇-含0.5%甲酸和0.5%三乙胺的水溶液;梯度:在0~90 min内甲醇从26%线性升至70%;流速:1 mL/min;检测波长:270 nm;柱温:30 ℃;进样量:5 μL。
2.2 溶液的配制
2.2.1 复方供试品溶液的制备 按处方配比精密取葛根芩连汤各味药饮片(葛根约15 g,黄芩约9 g,黄连约9 g,甘草约6 g),加10倍量水煎煮45 min,滤过,再加8倍量水煎煮30 min,减压回收干燥,得提取物。取提取物约0.5 g,精密称定,加甲醇25 mL,超声提取后经0.45 μm微孔滤膜滤过,得复方供试品溶液。
2.2.2 药材供试品溶液的制备 按处方量各取葛根、黄芩、黄连药材,精密称定,按复方相同制备方法处理,得各药材提取物,取葛根药材提取物约2 mg、黄芩药材提取物约1 mg、黄连药材提取物约1 mg,精密称定,各加甲醇10 mL,经0.45 μm微孔滤膜滤过,得各药材供试品溶液。
2.2.3 阴性供试品溶液的制备 称取除某味药材的阴性处方,按复方相同制备方法处理,得各阴性样品提取物,各取葛根、黄芩、黄连阴性提取物约3 mg,精密称定,加甲醇10 mL,经0.45 μm微孔滤膜滤过,得各阴性供试品溶液。
2.2.4 对照品溶液配制 精密称取经五氧化二磷干燥48 h的各对照品适量,用甲醇溶解并定容,制备各对照品的单标对照品溶液:葛根素浓度为0.35 mg/mL、大豆苷浓度为0.13 mg/mL、大豆苷元浓度为1.02 mg/mL、黄芩苷浓度为0.0712 mg/mL,汉黄芩苷浓度为0.18 mg/mL,黄芩素浓度为0.36 mg/mL,汉黄芩素浓度为0.14 mg/mL,盐酸小檗碱浓度为0.46 mg/mL,盐酸巴马汀浓度为0.52 mg/mL,备用。
2.3 指纹图谱方法学考察
2.3.1 对照品的检出 在确定的色谱条件下对葛根芩连汤中的葛根素、大豆苷、大豆苷元、黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、盐酸小檗碱、盐酸巴马汀均能检出,峰形均较理想(见图1)。
2.3.2 精密度试验 取同一供试品溶液,按上述色谱条件连续进样5次,记录各组分的保留时间和峰面积,各组分保留时间的RSD均小于0.71%,各组分峰面积的RSD均小于0.58%,结果表明仪器精密度良好。
2.3.3 稳定性试验 取同一供试品溶液,分别于0、2、4、6、12、24、36 h在上述色谱条件下进样分析,记录各组分的保留时间和峰面积,各组分保留时间的RSD均小于1.29%,各色谱峰峰面积的RSD均小于0.98%,结果表明供试品溶液在36h内稳定(见图2)。
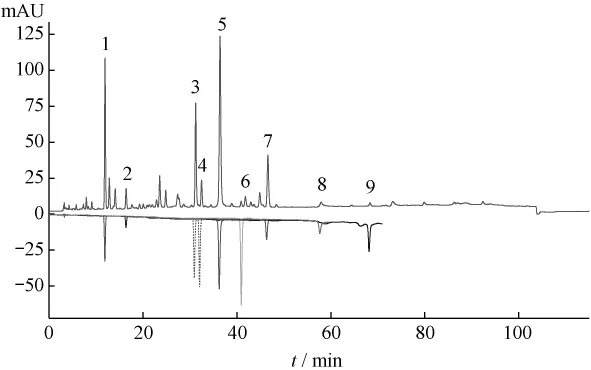
图1 葛根芩连汤HPLC图中对照品的检出
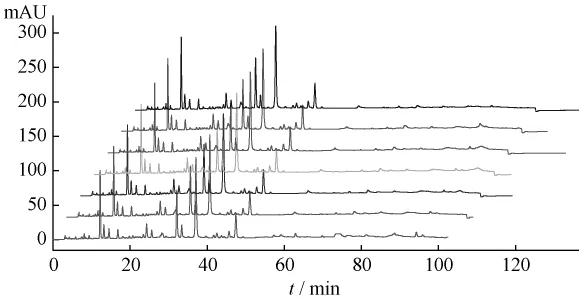
图2 不同时间间隔测定所得HPLC图
2.3.4 重复性试验 按处方配比取葛根芩连汤各味药饮片,各取5份,按2.2.1项方法制备样品,在上述色谱条件下进样分析,记录各组分的保留时间和峰面积,各组分保留时间的RSD均小于1.41%,各组分峰面积的 RSD均小于1.71%,结果表明本试验重复性良好。
2.4 HPLC相对保留值指纹谱的建立 本研究采用HPLC技术对葛根芩连汤及葛根、黄芩、黄连药材一次进样进行梯度分离分析,得到众多的色谱峰(见图3~5),利用相对保留值(α)及样品峰相对面积值(RA)组成各样品的HPLC数字化色谱指纹谱(HPLC-FPS)[3-6],通过与阴性样品比较,可找出葛根芩连汤中用以鉴定上述药材的较全面的特征峰群,实现对制剂中药材的鉴定。表1为以葛根素为内参照峰的葛根药材及样品的色谱指纹谱的特征峰;表2为以黄芩苷为内参照峰的黄芩药材及样品的色谱指纹谱的特征峰;表3为以盐酸小檗碱为内参照峰的黄连药材及样品的色谱指纹谱的特征峰。
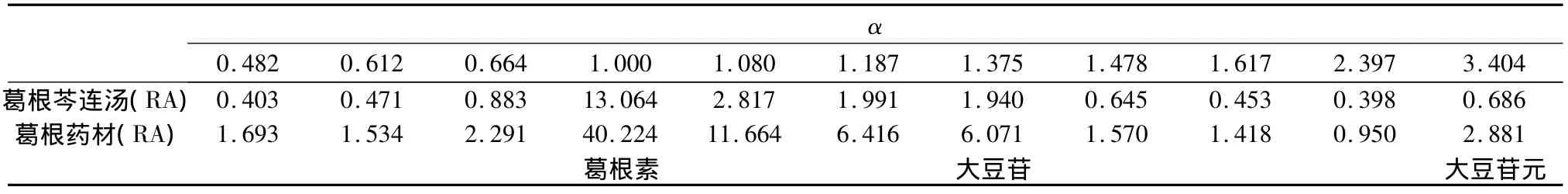
表1 葛根药材在样品中的特征峰

表2 黄芩药材在样品中的特征峰

表3 黄连药材在样品中的特征峰

图3 葛根芩连汤、葛根药材及阴性样品HPLC图
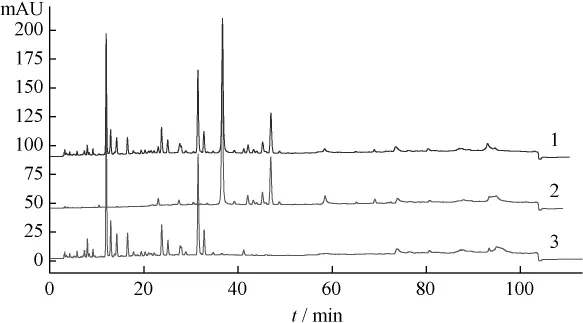
图4 葛根芩连汤、黄芩药材及阴性样品HPLC图

图5 葛根芩连汤、黄连药材及阴性样品HPLC图
2.5 结果 在葛根芩连汤指纹谱中均能找到葛根、黄芩和黄连药材的特征峰群,其中 α 值为0.482、0.612、0.664、1.000、1.080、1.187、1.375、1.478、1.617、2.397 和 3.404(以葛根素为内参照峰)为葛根药材的特征峰群;α值为0.633、0.753、1.000、1.070、1.147、1.179、1.232、1.279、1.595、1.884和1.998(以黄芩苷为内参照峰)为黄芩药材的特征峰群;α 值为 0.132、0.292、0.438、0.645、0.798、0.879、1.000、1.040、2.935和2.986(以盐酸小檗碱为内参照峰)为黄连药材的特征峰群。结果表明所建立的数字化色谱指纹谱中可找出用以鉴定各药材的较全面的特征峰群,可实现对复方中中药材的鉴定。
3 讨论
葛根芩连汤中成分复杂,含有多种生物碱、黄酮类、皂苷类成分,方中的黄酮类和生物碱类物质分别进行反相高效液相色谱分析时所适用的流动相条件差别较大。本研究考察了甲醇-水,甲醇-0.5%甲酸,甲醇-水(含0.5%甲酸和0.5%三乙胺)以及乙腈-水(含0.5%甲酸和0.5%三乙胺)等流动相对样品组分分离的影响,确定了最佳流动相配比。同时比较了在 210,220,250,270,280,346,365 nm 不同检测波长下的图谱,结果显示在270nm波长下,峰数目较多,各峰高低比例适宜,且基线平稳。建立的色谱条件可实现弱酸性和弱碱性物质同时分析,且在很大程度上解决了弱酸性和弱碱性物质共存时,弱碱性物质峰形差的问题。
药材来源的峰在复方中的表达可以划分一定的指纹区域,其中5~20 min主要来自葛根,20~35 min主要来自黄连,35~70 min主要来自黄芩,直观的指纹图谱是评价、控制中成药质量的有效方法,也是中药研究的一种模式。但这样的方法有一定误差且无量化指标。目前由于对照品有限,不能较全面地对指纹图谱中的主要色谱峰一一定性,本研究通过分析复方、待鉴药材和阴性样品的数字化色谱指纹谱,找到了葛根、黄芩和黄连的特征峰群,可用于复方中上述药味的鉴别,这也为今后葛根芩连汤化学配伍研究奠定了基础。
通过对所建立的数字化色谱指纹谱分析,发现在全方中保留时间为57.285、81.309、88.250 min为葛根、黄芩和黄连药材及各自的阴性样品中均未检测到的;提示上述成分为复方形成过程中产生的新成分,为进一步深入研究上述问题,需在中医理论指导下,设计不同配伍的药味在确定色谱条件下分析,建立相应的数字化色谱指纹谱进行更为深入地研究,如能结合液-质联用等技术,则有望搞清指纹谱中相关成分的结构,从而为阐明其药效物质基础提供科学依据。
本研究在确定的色谱条件下对炙甘草进行如上述药材的鉴定,结果不理想,炙甘草药材出峰非常少,在全方中只检出了保留时间为20.896 min处的甘草苷,含量非常低,未检测到甘草酸。笔者将炙甘草单独用甲醇提取后在确定色谱条件分析,可检出成分较在全方中多,分析原因可能是全方水煎后引起炙甘草中成分变化,有文献报道葛根芩连汤中黄连可降低其中甘草酸含量[7]。
[1]李顺保.伤寒论版本大全[M].北京:学苑出版社,2001.
[2]刘 斌,石任斌,朱丽君,等.苦参汤黄酮类成分HPLC指纹图谱及其与组方药味黄芩和苦参的相关性研究[J].中国中药杂志,2007,32(16):1631.
[3]洪筱坤.中药数字化色谱指纹谱[M].上海:上海科学技术出版社,2003.
[4]王新宏,安 睿,邹 云,等.数字化色谱指纹谱技术在双黄连制剂质量及药材鉴定中的应用[J].中草药,2002,33(2):115.
[5]安 睿,王新宏,周思丽,等.HPLC测定不同产地丹参药材中脂溶性成分分析[J].中成药,2004,26(4):249.
[6]安 睿,王新宏,唐 莹,等.不同产地丹参药材中水溶性成分分析[J].中成药,2005,27(7):812.
[7]戴开金,罗佳波,吴昭晖,等.配伍对葛根芩连汤中甘草酸含量的影响[J].中草药,2003,34(12):1084.
猜你喜欢
中医药导报(2022年3期)2022-11-09
中国药物经济学(2022年3期)2022-04-19
中国兽药杂志(2019年4期)2019-05-15
中成药(2018年5期)2018-06-06
天然产物研究与开发(2018年4期)2018-05-07
中成药(2017年5期)2017-06-13
中兽医学杂志(2016年2期)2016-01-31
中医研究(2014年10期)2014-03-11
中成药(2014年10期)2014-02-28