
乙炔氢氯化反应高分散载金催化剂的制备及催化性能
2010-01-29 02:10王声洁沈本贤肖卫国宋庆雷
石油学报(石油加工) 2010年2期
王声洁,沈本贤,肖卫国,宋庆雷
(1.华东理工大学化学工程联合国家重点实验室,上海200237;2.天津大沽化工股份有限公司,天津300455)
乙炔氢氯化反应合成氯乙烯单体,是一种最早工业化的氯乙烯生产工艺,在中国一直占有重要地位。但是该工艺采用剧毒且易升华损失的氯化汞触媒,不仅严重危害人体健康、污染环境,而且影响氯乙烯单体的产品质量。如何降低氯乙烯生产过程中所带来的触媒污染,以更好地适应国家建设资源节约型、环境友好型社会的要求,已成为整个电石乙炔法 PVC行业面临的一个亟待解决的问题。
多年来,人们曾致力于非汞触媒的研发,以期代替汞触媒[1-10],但都因活性低于汞触媒或者使用寿命太短而没能成功实现工业化应用。Hutchings等[5-10]的研究成果在该研究领域较有代表性。他们通过大量的筛选实验,总结出了催化乙炔氢氯化反应的金属活性规律,即金属的标准电极电势越高活性越高[5]。并根据该规律,以 HAuCl4为活性前驱体制备了用于乙炔氢氯化反应的Au/C催化剂。该催化剂虽具有比汞触媒更高的初活性,但是使用稳定性仍然欠佳,且制备催化剂时需采用王水作溶剂,存在较大的腐蚀及污染问题。迄今为止,虽然 Au催化领域的研究日益升温,但是关于Au催化乙炔氢氯化反应的研究在 Hutchings之后鲜有人涉及。
Au(en)2Cl3常被用作制备负载型高分散 Au催化剂的前驱体[11-15]。在 Hutchings研究的基础上,笔者采用 Au(en)2Cl3为前驱体,活性炭(AC)为载体,用等体积浸渍法制备了用于乙炔氢氯化反应的负载型Au催化剂。该制备工艺简单,无腐蚀及污染问题。在常压固定床反应器中对自制Au(en)2Cl3/AC催化剂催化乙炔氢氯化反应性能进行了研究,并考察了各种制备条件的影响,取得了有意义的研究结果,可为乙炔氢氯化反应非汞催化剂的开发提供参考。
1 实验部分
1.1 原料及试剂
乙炔,纯度99.99%,上海成功气体公司产品; HCl,纯度99.5%,北京华宇同方科技开发有限公司产品;椰壳质活性炭(ACS)和煤质活性炭(ACC), 20~40目,堆积密度0.48 g/cm3,上海活性炭厂产品;活性氧化铝(Al2O3),国药集团化学试剂有限公司产品;二氧化硅(SiO2),上海凌峰化学试剂有限公司产品;5A分子筛,自制;氯金酸(HAuCl4·4H2O),分析纯,国药集团化学试剂有限公司产品;实验所用其他药品均为分析纯。
1.2 Au(en)2Cl3的合成[16]
将 HAuCl4·4H2O(1.0 g)溶于10 mL去离子水中,慢慢加入0.45 mL无水乙二胺,溶液变为红棕色;剧烈搅拌30 min后,加入70 mL乙醇溶液,立刻产生白色沉淀;将该悬浮液再搅拌20 min后,过滤,白色沉淀用无水乙醇洗涤,最后放于真空干燥箱中40℃干燥24 h,备用。
1.3 催化剂的制备
配制不同浓度的Au(en)2Cl3溶液作为Au前体溶液,采用等体积浸渍将 Au(en)2Cl3浸渍到活性炭上,在空气中自然晾干后,于烘箱中80℃干燥24 h,即得新鲜催化剂样品,标记为Au/AC。分别配制一定浓度的 NaCl、KCl和MgCl2的溶液等体积浸渍于Au/AC催化剂上,放于烘箱中80℃干燥24 h,可得碱金属修饰的Au-Na/AC、Au-K/AC和Au-Mg/AC催化剂。将 Au/AC催化剂在 H2气氛下,于300℃还原2 h,得还原后的 Au(R)/AC催化剂。
Au/Al2O3、Au/SiO2和 Au/5A分子筛催化剂的制备方法同Au/AC。催化剂样品的Au负载量均以金属质量计。
1.4 催化剂活性评价
采用内径10 mm的不锈钢常压流动固定床反应器,以乙炔氢氯化反应评价催化剂的催化活性。反应器采用循环油浴控温,催化剂装填量5 g。乙炔经装有 FeCl3的净化器除去硫、磷、砷等杂质,再经装有硅胶的干燥器脱除水分;HCl气体经5A分子筛吸附柱干燥。采用质量流量计控制气体流量。反应前用N2吹扫除去系统中水分和空气,然后通入乙炔和 HCl 2种原料气进行反应。反应产物经NaOH吸收除去 HCl气体之后,进行色谱分析。
1.5 分析方法及评价指标
采用上海海欣色谱仪器有限公司 GC-920气相色谱仪分析反应物及产物组成。色谱仪配以氢火焰离子化检测器和 PLOT Al2O3色谱柱。PLOT Al2O3色谱柱由中国科学院兰州化学物理研究所研制,AT.Al2O3/S固定液,柱长50 m,内径0.53 mm,膜厚20.0μm。
分析条件:柱温50℃,检测器温度180℃,汽化室温度180℃,进样量80μL。
用乙炔的转化率 xA和氯乙烯的选择性 sVC来评价催化剂的活性。因反应后氯化氢被吸收除去,计算时可把整个反应体系看作体积不变,总体积按1个体积单位计算,公式如下:

式(1)、(2)中,φA1为剩余乙炔的体积分数;φVC为氯乙烯的体积分数。
1.6 催化剂的表征
采用日本理光机电公司Rigaku D/max 2550VB/PC X射线衍射仪进行样品的XRD分析。管电压40 kV,管电流 100 mA,Cu靶。采用美国 Micromeritics Instrument Corporation ASAP2010型微结构分析仪测定样品的比表面积。190℃下脱气6 h、77 K下进行液氮吸附。采用J EOL J EM-100 CXⅡ透射电镜观察样品的粒径分布,加速电压为100 kV。
2 结果与讨论
2.1 Au/AC催化剂制备条件对其催化活性的影响
2.1.1 Au价态的影响
以椰壳质活性炭(ACS)为载体制备了 Au/ACS催化剂,对 H2还原前后的催化剂 Au/ACS和Au(R)/ACS进行了 XRD表征,结果示于图1。从图1可见,H2还原前的Au/ACS的XRD谱中,除活性炭的非晶态衍射峰之外,无其他晶态衍射峰出现,表明Au/ACS的活性组分在活性炭表面高度分散或呈非晶态分散于活性炭表面;经 H2还原后, Au(R)/ACS在2θ为38.12、44.34、64.52和77.66°处出现典型Au0衍射峰,表明此时活性组分主要为Au0。在相同条件下,Au/ACS和Au(R)/ACS分别用于催化乙炔氢氯化反应的催化活性示于图2。由图2可知,Au/ACS催化乙炔氢氯化反应具有较长的诱导期,催化活性(乙炔转化率)随时间的延长逐渐增大;在反应开始约10 h后,催化剂的活性趋于稳定。这是由于前驱体 Au(en)2Cl3具有较大的空间位阻,反应气体克服空间位阻及能级壁垒与中心Au离子配位形成具有催化活性的过渡态产物需要一个较为缓慢的过程。而Au(R)/ACS催化乙炔氢氯化反应时,反应开始1 h,乙炔转化率约65%,之后随时间延长逐渐降低。由此可见,Au/ACS催化剂中的 Au还原为 Au0后,其催化活性明显降低。Nkosi等[8]的研究已证实,载 Au催化剂催化乙炔氢氯化反应的活性从高到低按其中 Au价态排列的顺序为Au3+、Au1+和 Au0。因此可见,自制Au/AC催化剂中 Au大部分以离子态形式 Aun+(Au3+或Au1+)存在,该离子态的Aun+为Au/AC催化乙炔氢氯化反应的活性中心。


2.1.2 载体的影响
在相同条件下,考察了活性 Al2O3、SiO2、5A分子筛、煤质活性炭(ACC)和椰壳质活性炭(ACS) 5种不同载体对负载 Au催化剂乙炔氢氯化反应催化活性的影响,结果列于表1。由表1可见,在催化乙炔氢氯化反应中,上述5种不同载体负载 Au催化剂均具有一定活性,其中以活性炭作载体时的活性最高。而在2种活性炭中,又以比表面积相对较高的椰壳质活性炭作载体的载 Au催化剂Au/ACS活性最高。这可能是由于椰壳质活性炭具有丰富的孔道结构和更高的比表面积,活性组分在其表面可以充分分散,有利于气相反应物与活性位的广泛接触的缘故。
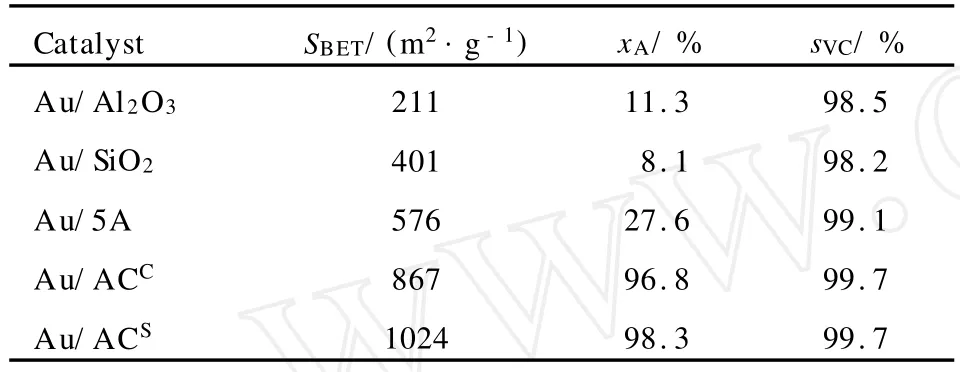
表1 不同载体的载Au催化剂催化乙炔氢氯化反应的转化率(xA)和氯乙烯选择性(sVC)Table 1 Reaction conversion(xA)and selectivity of vinyl chloride(sVC)in acetylene hydrochlorination catalyzed by Au-supported catalyst with different supports
2.1.3 Au负载量的影响
以椰壳质活性炭为载体,制备了一系列不同负载量的Au/ACS催化剂。在相同反应条件下,考察了它们在乙炔氢氯化反应中的催化活性,结果示于图3。
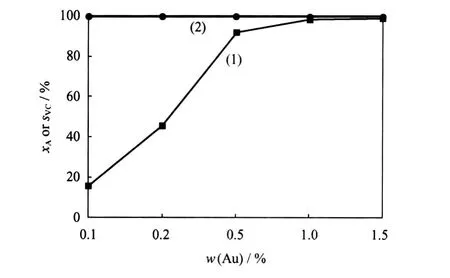
图3 不同Au负载量的Au/ACS催化乙炔氢氯化反应的转化率(xA)和氯乙烯选择性(sVC)Fig.3 Reaction conversion(xA)and selectivity of vinyl chloride(sVC)in acetylene hydrochlorination catalyzed by Au/ACSwith different Au loadingsReaction condition:T=180℃;GHSV=240 h-1; V(HCl)/V(C2H2)=1.1;t=10 h(1)xA;(2)sVC
由图3可见,当Au负载质量分数由0.1%增加到1.0%时,Au/ACS催化剂的催化活性逐渐提高,乙炔转化率由15.6%增加至98.3%,氯乙烯选择性均在 99.5%以上。由于 Au负载量的增加, Au/ACS催化剂表面的活性位逐渐增多,因此催化剂的活性逐渐提高;当 Au的负载质量分数大于1.0%时,催化剂的活性随Au负载量增加而提高的趋势不再明显。因此,乙炔氢氯化的Au/AC催化剂中Au的负载质量分数以1.0%为宜。
2.1.4 碱金属助剂的影响
碱金属常作为电子助剂加入到固相催化剂中。通过向1%(质量分数)Au/ACS催化剂中添加适量NaCl、KCl、MgCl2助剂,合成了 Au-Na/ACS、Au-K/ACS、Au-Mg/ACS催化剂。考察了碱金属对Au/ACS催化剂乙炔氢氯化反应催化活性的影响,结果见图4。
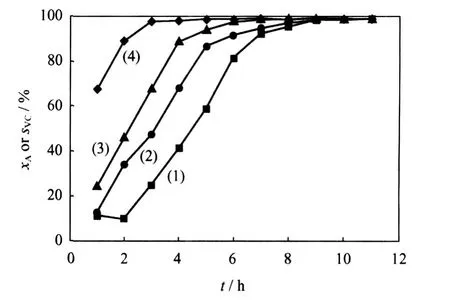
图4 含不同碱金属助剂的 Au/ACS催化乙炔氢氯化反应的转化率(xA)和氯乙烯选择性(sVC)Fig.4 Reaction conversion(xA)and selectivity of vinyl chloride(sVC)in acetylene hydrochlorination catalyzed by Au/ACScontaining different alkali metals Reaction condition:T=180℃;GHSV=240 h-1;V(HCl)/V(C2H2)=1.1;w(Au)=1.0%; w(Na)=w(Mg)=w(K)=0.5%(1)Au/ACS;(2)Au-Na/ACS;(3)Au-Mg/ACS;(4)Au-K/ACS
由图4可见,碱金属助剂的加入,不同程度地缩短了 Au/ACS催化剂催化乙炔氢氯化反应诱导期,而对反应转化率和氯乙烯选择性近乎没有影响。在所考察的碱金属助剂中,K缩短反应诱导期的效果最为明显,这可能是由于碱金属 K无 d电子和d空穴,具有较强的给电子能力[17-19],它的加入使得Au周围的电子云密度增加,从而更易于吸附反应物,起到了活化活性炭表面Au的作用。
当 K的添加量较低时(质量分数0.2%),并不能明显缩短 Au/ACS催化剂催化乙炔氢氯化反应诱导期,而当 K的添加质量分数为0.5%时,诱导期明显缩短;过多的 K添加量(质量分数2.0%)则会造成活性中心被覆盖,催化剂活性下降,如图5所示。

图5 不同 K添加量的Au-K/ACS催化乙炔氢氯化反应的转化率(xA)和氯乙烯选择性(sVC)Fig.5 Reaction conversion(xA)and selectivity of vinyl chloride(sVC)in acetylene hydrochlorination catalyzed by Au-K/ACSwith different KamountsReaction condition:T=180℃;GHSV=240 h-1; V(HCl)/V(C2H2)=1.1;w(Au)=1.0%w(K)/%:(1)0.2;(2)0.5;(3)1.0;(4)2.0
2.2 优化制备条件下Au/AC催化剂的催化活性
众多研究[20-23]表明,Au的催化活性与其粒径大小密切相关,粒径越小活性越高。笔者采用Au(en)2Cl3为前驱体,椰壳质活性炭(ACS)为载体, KCl为助剂,制备了 Au-K/ACS催化剂。图6、7分别为Au-K/ACS的 TEM照片和粒径分布图。由图6、7可以看出,Au-K/ACS催化剂具有较高分散性,平均粒径仅为2.3 nm,与 Conte等[10]所制备的乙炔氢氯化反应Au催化剂的平均粒径(4.8 nm)相比,减小了1/2。不同反应条件下新鲜Au-K/ACS催化剂用于乙炔氢氯化反应的转化率示于图8。由图8可见,在一定空速和反应配比下,升高温度有利于Au-K/ACS催化活性的提高;在150℃时,乙炔转化率为91%左右,180℃时的乙炔转化率可达98%以上,氯乙烯选择性可达99.5%以上,与目前汞触媒的活性相当。在20倍于目前汞触媒的空速下(GHSV=2000 h-1)反应20 h,Au-K/ACS催化剂活性平稳;反应后的催化剂平均粒径为3.2 nm,相比新鲜催化剂粒径,无明显增大。目前工业上应用的汞触媒在高空速下会因汞的升华损失而迅速失活,从而限制了其生产能力。而 Nkosi等[8]以 HAuCl4制备的载Au催化剂虽无活性组分流失问题,但是在高空速下操作,6 h内催化活性会明显下降。由此可见,自制Au-K/ACS催化剂具有较好的催化活性稳定性。
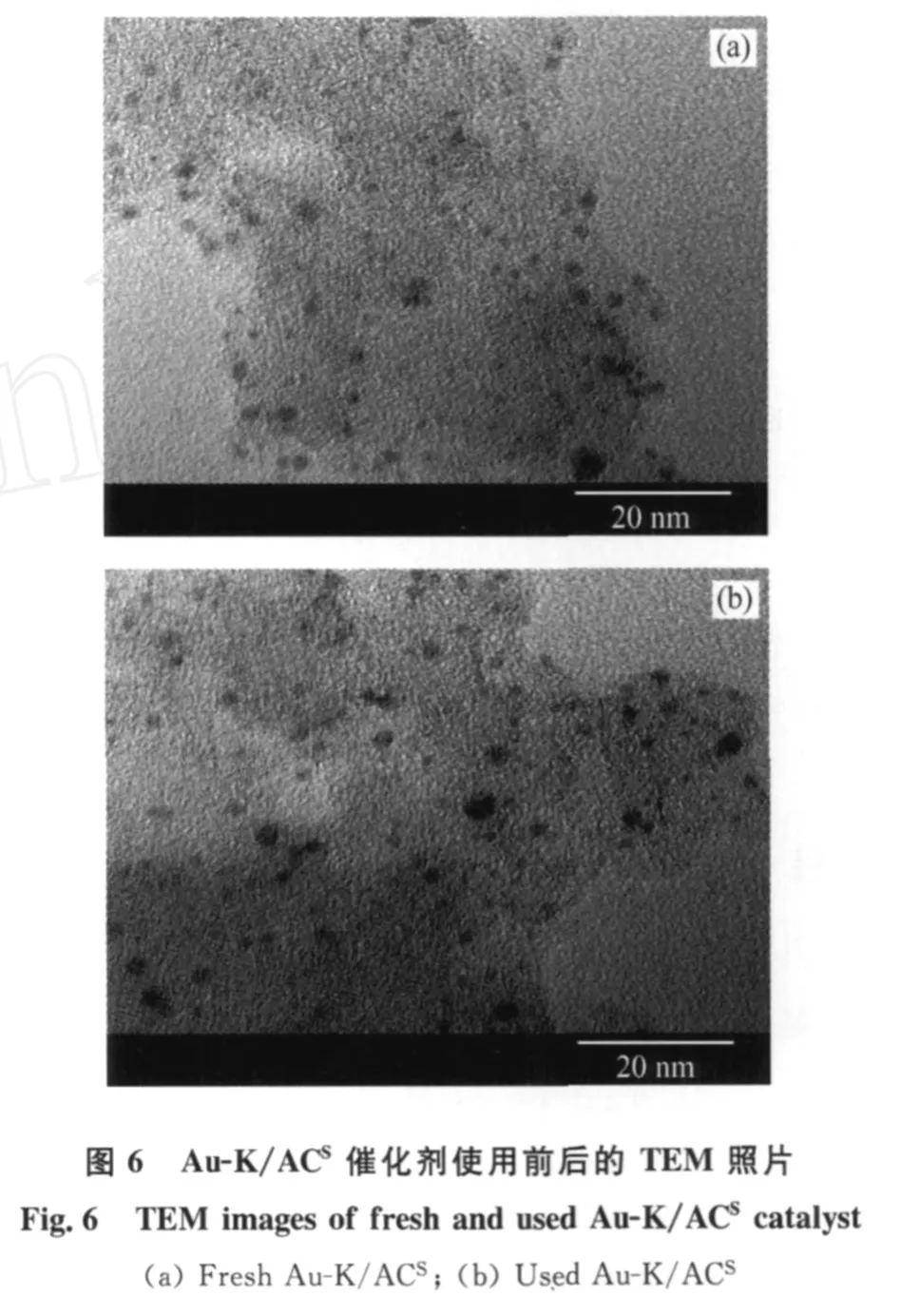
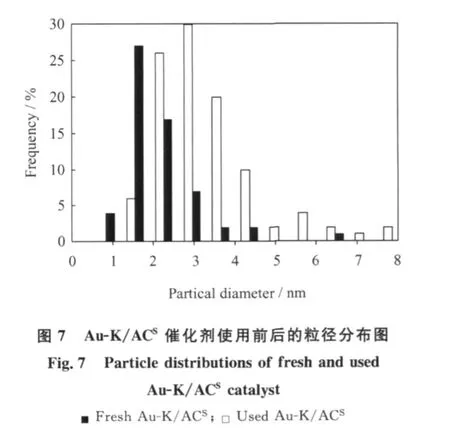

图8 不同反应条件下Au-K/ACS催化乙炔氢氯化反应的转化率(xA)Fig.8 Reaction conversion(xA)of acetylene hydrochlorination catalyzed by Au-K/ACSunder different reaction conditionsw(Au)=1.0%;w(K)=0.5%(1)T=150℃,GHSV=240 h-1,V(HCl)/V(C2H2)=1.1;(2)T=180℃,GHSV=240 h-1,V(HCl)/V(C2H2)=1.1;(3)T=180℃,GHSV=2000 h-1,V(HCl)/V(C2H2)=1.0
3 结 论
(1)以 Au(en)2Cl3为活性前驱体制备了用于乙炔氢氯化反应的载Au催化剂。采用椰壳质活性炭(ACS)为载体,Au负载质量分数为 1%时, Au/ACS催化剂具有较高的催化活性,但诱导期较长。加入 KCl助剂(K质量分数0.5%)可有效缩短Au/ACS催化剂的诱导期。
(2)Au/AC催化剂中Au大部分以离子态形式Aun+(Au3+或Au1+)存在,该离子态的 Au活性组分为Au/AC催化乙炔氢氯化反应的活性中心。
(3)以 KCl为助剂的Au-K/ACS催化剂具有较高分散性,平均粒径仅为2.3 nm。在温度为180℃、空速为240 h-1、V(HCl)/V(C2H2)=1.1时,该催化剂催化乙炔氢氯化反应得乙炔转化率可达98%以上,氯乙烯选择性不低于99.5%,且在空速为2000 h-1,仍具有较好的稳定性。
[1]邓国才,吴本湘,李同树,等.乙炔法合成氯乙烯固相非汞催化剂的研制[J].聚氯乙烯,1994,(6):5-9. (DENG Guocai,WU Benxiang,LI Tongshu,et al. Preparation of solid phase non-mercuric catalysts for synthesis of vinyl chloride via acetylene method[J]. Polyvinyl Chloride,1994,(6):5-9.)
[2]杨琴,罗芩,蒋文伟,等.乙炔氢氯化反应气固相非汞催化体系研究[J].四川化工,2007,10(5):13-15. (YANG Qin,LUO Qin,J IANG Wenwei,et al.Studies on the vapor phase hydrochlorination system by use of mereury free catalysts[J].Sichuan Chemical Industry, 2007,10(5):13-15.)
[3]MITCHENKO S A,KHOMUTOV E V,SHUBIN A A, et al.Catalytic hydrochlorination of acetylene by gaseous HClon the surface of mechanically pre-activated K2Pt2Cl6salt[J].J Mol Catal A,2004,212(1):345-352.
[4]SOLVAY.Process forpreparing vinylchloride by reacting acetylene with hydrogen chloride: DE, 3824634A1[P].1988-7-20.
[5]HUTCHINGS G J.Vapor phase hydrochlorination of acetylene:Correlation of catalytic activity of supported metal chloride catalysts[J].J Catal,1985,96(1):292-295.
[6]HUTCHINGS G J. Gold catalysis in chemical processing[J].Catal Today,2002,72(1-2):11-17.
[7]NKOSIB, HUTCHINGS G J. Vapourphase hydrochloride of acetylene with groupⅧandⅠB metal chloride catalysts[J].Appl Catal,1988,43(1):33-39.
[8]NKOSI B,COVILLE N J,HUTCHINGS G J,et al. Hydrochlorination of acetylene using gold catalysts:A study of catalyst deactivation[J].J Catal,1991,128 (2):366-377.
[9]NKOSI B,ADAMS M D,COVILL E N J,et al. Hydrochlorination of acetylene using carbon-supported gold catalysts:A study of catalyst reactivation[J].J Catal,1991,128(2):378-386.
[10]CONTE M,CARL EY A F,HEIRENE C,et al. Hydrochlorination of acetylene using a supported gold catalyst:A study of the reaction mechanism[J].J Catal,2007,250(2):231-239.
[11]BULUSHEV D A,YURANOV I,SUVOROVA E I, et al.Highly dispersed gold on activated carbon fibers for low-temperature CO oxidation[J].J Catal,2004, 224(1):8-17.
[12]ZHU Haoguo,MA Zhen,CLARK J C,et al.Lowtemperature CO oxidation on Au/fumed SiO2-based catalysts prepared from Au(en)2Cl3precursor[J].Appl Catal A,2007,326(1):89-99.
[13]RIAHIA G, GUILL EMOTA D, POLISSETTHFOINA M,et al.Preparation,characterization and catalytic activity ofgold-based nanoparticles on HY zeolites[J].Catal Today,2002,72(1-2):115-121.
[14]GUILLEMOT D,POLISSET-THFOIN M,FRAISSARD J. Preparation of nanometeric gold particles on NaHY[J]. Catal Lett,1996,41(3-4):143-148.
[15]张竞杰,张彭义,张博,等.活性炭负载金催化分解空气中低浓度臭氧[J].催化学报,2008,29(4):335-340. (ZHANGJingjie,ZHANG Pengyi,ZHANGBo,et al. Decomposition of low-level ozone in air over activated carbon-supported gold catalyst[J].Chinese Journal of Catalysis,2008,29(4):335-340.)
[16]BLOCKB P,BAILAR J C.The reaction of gold(Ⅲ) with some bidentate coordinating groups[J].JAm Chem Soc,1951,73(10):4722-4725.
[17]夏明,何淡云,祝以湘.乙苯脱氢 Fe2O3-K2O系催化剂中晶格氧和钾的助催作用[J].分子催化,1995,9(3): 201-205.(XIA Ming,HE Danyun,ZHU Yixiang.The role of lattice oxygen and potassium in iron-oxide-based catalyst for dehydrogenation of ethylbenzene[J].J M Catal(China),1995,9(3):201-206.)
[18]LIU Jie,LU Xueju,ZHOU Guangdong,et al.Effect of KCl on CuCl2/γ-Al2O3catalyst for oxychlorination of ethane[J].React Kinet Catal Lett,2006,88(2):315-323.
[19]李玉敏.工业催化原理[M].天津:天津大学出版社, 1996:73.
[20]L EE S J,GAVRIILIDIS A.Supported Au catalysts for low-temperature CO oxidation prepared by impregnation [J].J Catal,2002,206(2):305-313.
[21]BOWKER M,NUHU A,SOARES J.High activity supported gold catalysts by incipient wetness impregnation[J].Catal Today,2007,122(3-4):245-247.
[22]BOND G C,TBOMPSON D T. Gold-catalysed oxidation of carbon monoxide[J].Gold Bull,2000,33 (2):41-51.
[23]CHEN Y J,YEH C T.Deposition of highly dispersed gold on alumina support[J].J Catal,2001,200(1):59-68.
猜你喜欢
中国盐业(2018年16期)2018-12-23
科普创作(2018年1期)2018-11-30
劳动保护(2018年8期)2018-09-12
聚氯乙烯(2018年12期)2018-06-06
聚氯乙烯(2018年7期)2018-02-18
中国氯碱(2017年2期)2017-03-13
电源技术(2016年9期)2016-02-27
自动化博览(2014年8期)2014-02-28
中国氯碱(2014年11期)2014-02-28
中国氯碱(2014年10期)2014-02-28
