
基于SVM机器学习筛选多发性骨髓瘤的特征基因及免疫浸润分析
2025-02-24 00:00:00石蕾张红宾
重庆医科大学学报 2025年1期
【摘 要】目的:应用生物信息学技术探究多发性骨髓瘤(multiple myeloma,MM)的遗传异质性以及免疫细胞在其病理生理中发挥的重要调节作用。方法:通过美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)公共高通量基因表达(gene expression omnibus,GEO)数据库对MM相关的数据集GSE125364、GSE72213采用生物信息学及机器学习等方法筛选MM诊断的关键基因,探索MM差异基因相关通路计算免疫细胞浸润情况,并通过分子生物学实验进行验证。结果:基于公共数据库的多发性骨髓瘤基因芯片数据采用生物信息学方法分析筛选差异基因410个,其中MM患者较对照组下调259个,上调151个。通过GO富集分析发现差异基因主要参与生物学过程包括DNA复制、染色体分离及有丝分裂;细胞定位主要富集在染色体区、纺锤体;分子功能主要富集在单链DNA螺旋酶活性,作用于DNA的催化和依赖ATP的活性等。KEGG通路富集分析显示的主要信号通路包括细胞周期、p53信号通路、细胞衰老和DNA复制等。GSEA分析对照组主要富集细胞周期、DNA复制、嘌呤代谢及核糖体等通路,MM组主要富集脂肪细胞因子信号通路、细胞粘附分子、核糖核酸多聚酶及抗坏血酸和醛酸代谢等通路。通过支持向量机递归特征消除(support vector machine-recursive feature elimination,SVM-RFE)算法筛选出2个MM诊断基因CPXM1和UROD,通过CIBERSORTx进行了免疫浸润分析表明CPXM1和UROD与免疫浸润相关,并通过MM.1S细胞进行了qRT-PCR验证(P<0.05)。结论:采用生物信息学方法能够有效分析MM与正常对照人群差异表达的基因,本次研究筛选出多发性骨髓瘤诊断的关键基因CPXM1和UROD,其表达与免疫浸润相关,可作为后续MM基础和临床实验研究的新靶点。
【关键词】多发性骨髓瘤;生物标志物;生物信息学;免疫浸润
【中图分类号】R733.3【文献标志码】A【收稿日期】2024-05-14
Screening for feature genes and immune infiltration of multiple myeloma: a study based on support vector machine
Shi lei,Zhang Hongbing
(Department of Hematology,The First Affiliated Hospital of Chongqing Medical University)
【Abstract】Objective:To investigate the genetic heterogeneity of multiple myeloma(MM) and the important regulatory role of immune cells in its pathophysiology by using bioinformatics techniques. Methods:The datasets of GSE125364 and GSE72213 associated with MM were obtained from the gene expression omnibus database of National Center for Biotechnology Information,and bioinformatics and machine learning methods were used to identify the key genes for the diagnosis of MM. Pathways associated with the differentially expressed genes in MM were analyzed to calculate immune cell infiltration,and molecular biology experiments were used for validation. Results:In this study,a total of 410 differentially expressed genes were obtained by the bioinformatics methods based on the gene mi‐croarray data of MM from public databases,among which 259 were downregulated and 151 were upregulated in MM patients compared with controls. The gene ontology enrichment analysis showed that the differentially expressed genes were mainly involved in the biologi‐cal processes such as DNA replication,chromosome segregation,and mitosis; as for cellular localization,they were mainly enriched in chromosomal region and the spindle apparatus; as for molecular function,they were mainly enriched in single-stranded DNA helicase activity,DNA catalysis,and ATP-dependent activity. The KEGG pathway enrichment analysis showed that the main signaling path‐ways included cell cycle,the p53 signaling pathway,cellular senescence,and DNA replication. The GSEA analysis showed that in the control group,the genes were mainly enriched in cell cycle,DNA replication,purine metabolism,and ribosomes,while in the MM group,the genes were mainly enriched in the adipokine signaling pathway,cell adhesion molecules,ribonucleic acid polymerase,and ascorbate and aldarate metabolism pathways. Two genes,CPXM1 and UROD,were obtained for the diagnosis of MM by support vector machine-recursive feature elimination algorithm,and the immune infiltration analysis via CIBERSORTx showed that CPXM1 and UROD were associated with immune infiltration; qRT-PCR validation was performed in MM.1S cells (P<0.05).Conclusion:Bioinfor‐matics methods can be used to effectively analyze the differentially expressed genes between MM patients and the normal control popu‐lation,and the key genes CPXM1 and UROD are obtained for the diagnosis of MM and are associated with immune infiltration,which can be used as new targets for subsequent basic and clinical experimental studies on MM.
【Key words】multiple myeloma;biomarkers;bioinformatics;immune infiltration
多发性骨髓瘤(multiple myeloma,MM)是一种起源于浆细胞的血液系统恶性肿瘤。其特征是骨髓中恶性浆细胞(plasma cell,PC)的克隆性增殖,伴随单克隆免疫球蛋白(称为M蛋白)的过度产生和终末器官损伤。主要临床表现包括高钙血症、肾脏损害、贫血及骨质破坏。MM是全球第二常见的血液系统恶性肿瘤,每年全球约有588 161例被确诊为多发性骨髓瘤,在美国每年约有3 490例确诊[1]。在过去的20年里,针对MM的治疗研发了许多新治疗方案,包括蛋白酶体抑制剂、免疫调节药物、单克隆抗体和CAR-T细胞疗法等[2]。随着这些新疗法的出现,MM5年相对存活率提高至54%[3]。但大多数MM患者仍会复发,而每次复发都会使得原有治疗变得更加困难[4]。以往研究发现免疫细胞参与了MM病程中的骨破坏和骨形成,且免疫细胞和骨细胞存在交互作用[5]。因此,探究MM的遗传异质性以及免疫细胞在其病理生理中发挥的重要调节作用将有助于对疾病预后进展以及治疗耐药机制提供更多的理论依据。通过美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)公共高通量基因表达(gene expression omnibus,GEO)数据库对MM相关的数据集采用机器学习、生物信息学以及分子生物学实验等方法筛选MM诊断的关键基因,探索MM相关基因通路并计算免疫细胞浸润,以求揭示MM内在病理生理机制并提供新的理论依据。
1 材料与方法
1.1 一般资料
本研究中MM相关资料来源于GEO数据库。通过对MM数据集进行筛选,选择含有正常对照和MM骨髓组织样本的数据集。GSE125364数据集中共有48个样本,其中3个正常样本,45个MM骨髓浆细胞样本,其作为训练集。GSE72213数据集中共有22个样本,其中3个正常样本,19个MM骨髓浆细胞样本,其作为验证集。
1.2 材料
MM细胞系MM.1S购自中国科学院上海细胞库。MM.1S细胞的培养采用RPMI 1640培养基,并加入10%热灭活胎牛血清(fetal bovine serum,FBS)及双抗(青霉素和链霉素),放置在5% CO?、37 °C饱和湿度培养箱中进行培养。实验取对数生长期的细胞进行。
1.3 方法
1.3.1 差异表达基因(differentially expressed genes,DEGs)分析 该部分使用R语言软件完成。调用R软件的“limma”包对GSE125364数据集进行差异分析。以|logFC|≥2.0和矫正后P<0.05为标准筛选DEGs,采用“pheatmap”及“ggplot2”绘制热图和火山图。
1.3.2 GO(Gene Ontology)/KEGG(Kyoto Encyclopedia of Genes and Genomes)/DO(Disease Ontology)/GSEA(Gene Set Enrichment Analysis)分析 该部分使用R软件完成。调用R软件的“clusterProfiler”“org.Hs.eg.db”“enrichplot”“DOSE”及“GSEABase”等R包对GSE125364数据集的样本进行分析。P值过滤条件为0.05,矫正后的P值过滤条件为0.05。
1.3.3 机器学习筛选诊断基因 将1.3.1分析得到差异表达基因进行机器学习,主要采用支持向量机-递归特征消除(support vector machine-recursive feature elimination,SVMRFE)算法,本部分调用R包“e1071”“kernlab”及“caret”完成SVM-RFE 算法对诊断基因的筛选。
1.3.4 受试者工作特征(receiver operating characteristic,ROC)曲线绘制 该部分使用R软件完成。调用R软件的“pROC”包对GSE72213数据集的样本进行分析。
1.3.5 免疫浸润分析 此部分通过cibersortx网站(https:// cibersortx.stanford.edu/)对GSE125364数据集进行分析,采用R软件中“corrplot”、“vioplot”及“ggpubr”等R包进行作图。
1.3.6 qRT-PCR验证 采用免疫磁珠分选正常骨髓CD138阳性浆细胞为对照细胞。将MM.1S细胞按照1×106/mL加入1 mL RNAiso Plus裂解试剂中,然后经过氯仿和异丙醇沉淀步骤,最终用乙醇清洗和溶解的方式提取RNA并测量其浓度;按照宝日医生物技术(北京)有限公司提供的试剂Prime‐Script? RT Reagent Kit with gDNA Eraser (Perfect Real Time)进行逆转录及扩增。引物序列见表1。

以2-ΔΔCt法计算目的基因的相对表达量;应用Graph‐Pad 9.0统计学软件对数据进行分析。
2 结果
2.1 DEGs分析
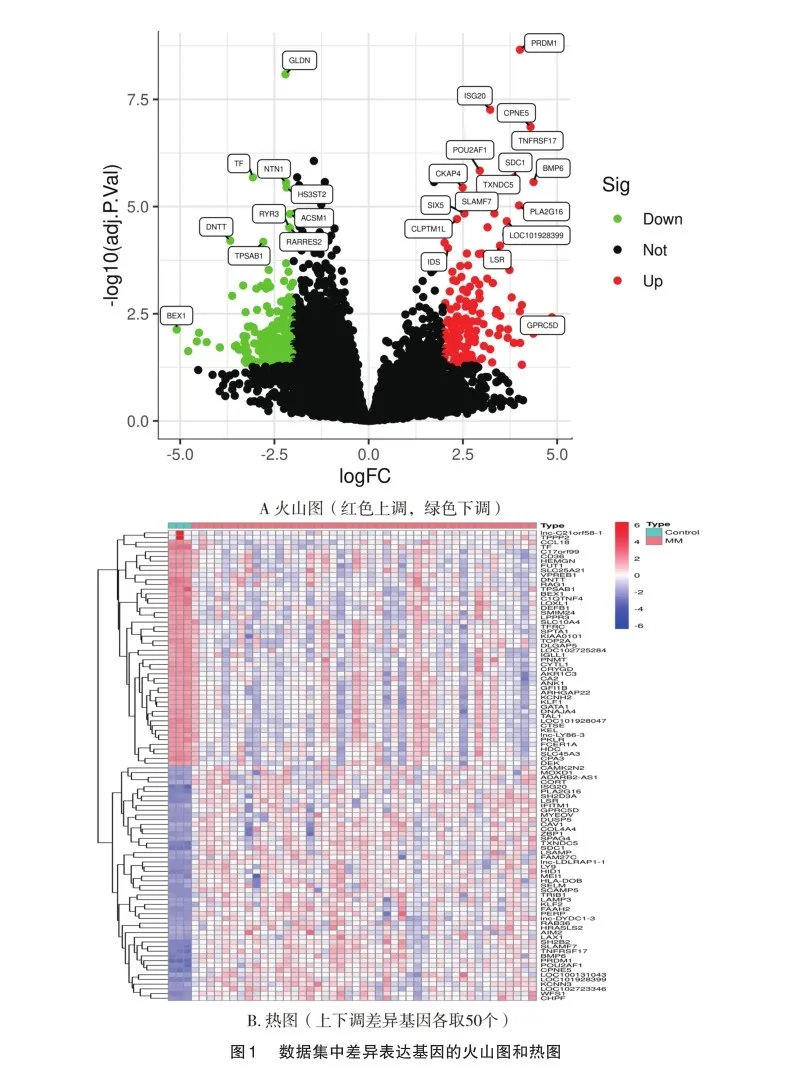
通过对GSE125364数据集进行差异分析后,根据筛选条件共得到差异基因410个,其中MM患者较对照组下调259个,上调151个(图1)。
2.2 GO(Gene Ontology)/KEGG(Kyoto Encyclopedia of Genes and Genomes)/DO(Disease Ontology)/GSEA(Gene Set Enrich‐ment Analysis)分析
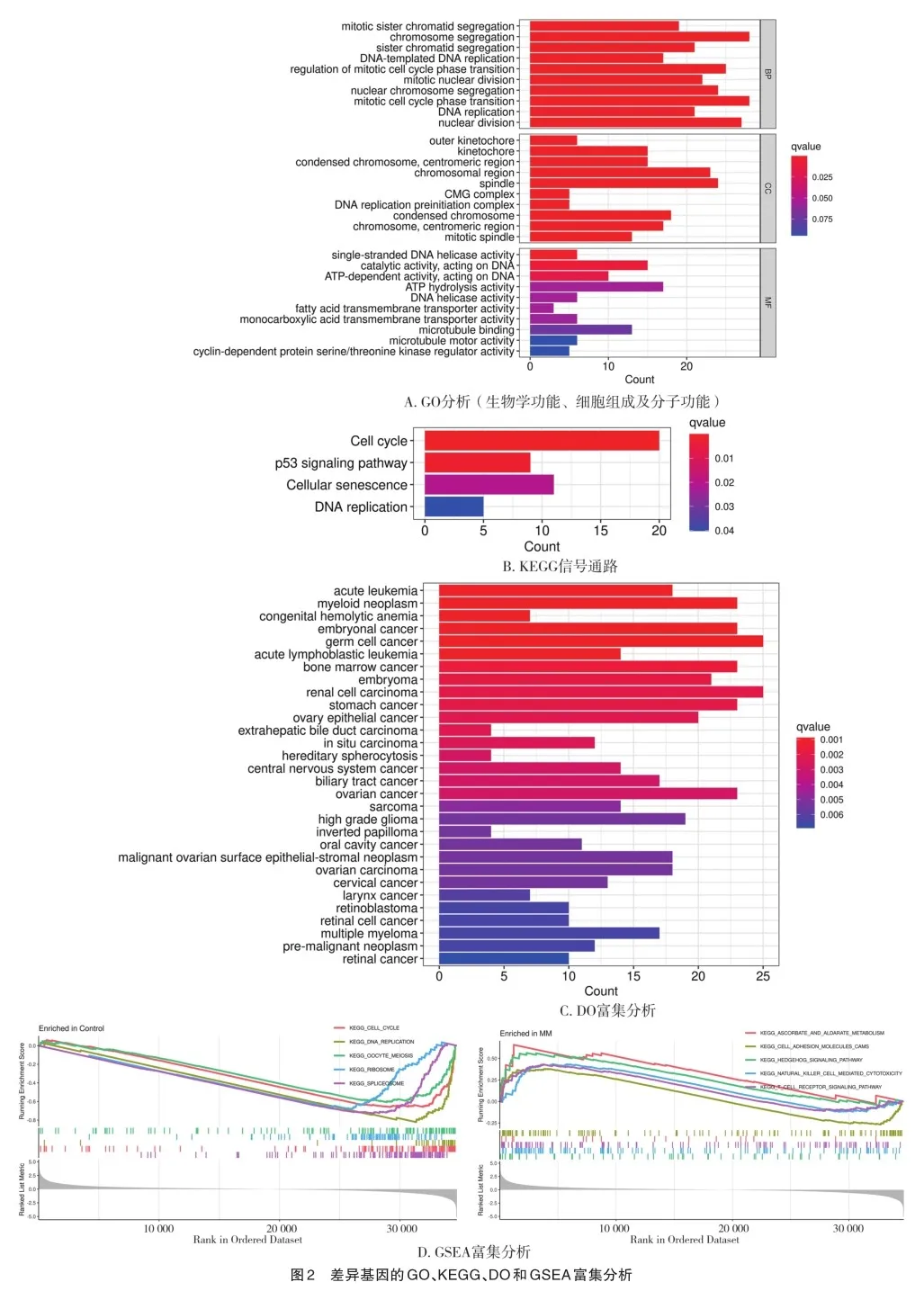
对筛选出的410个差异表达基因进行GO、KEGG通路富集、DO和GSEA分析显示,差异基因生物学过程(biological process,BP)主要富集在DNA复制、染色体分离及有丝分裂;细胞组成(cellular component,CC)主要富集在染色体区、纺锤体,分子功能(molecular function,MF)主要富集在单链DNA螺旋酶活性,作用于DNA的催化和依赖ATP的活性等(图2A);共富集到KEGG通路4个,主要涉及细胞周期、p53信号通路、细胞衰老和DNA复制(图2B);DO主要富集的疾病为急性白血病、骨髓肿瘤、急性淋巴细胞白血病及多发性骨髓瘤等(图2C);GSEA分析Control组主要富集细胞周期、DNA复制、嘌呤代谢及核糖体等通路,MM组主要富集脂肪细胞因子信号通路、细胞粘附分子、核糖核酸多聚酶及抗坏血酸和醛酸代谢等通路(图2D)。
2.3 机器学习筛选诊断基因及验证
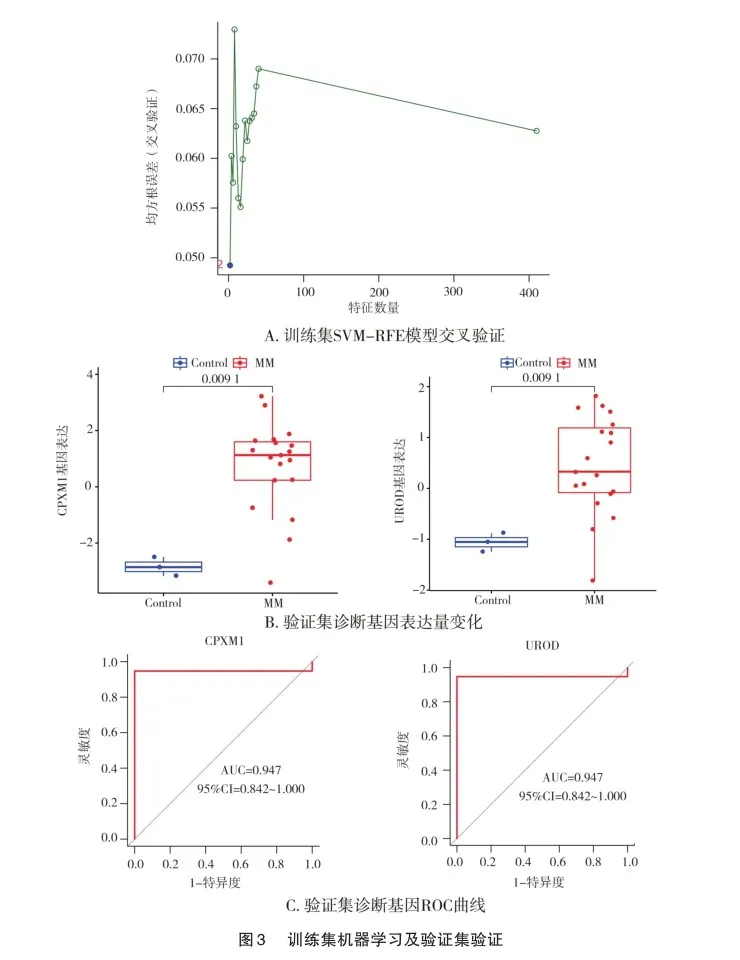
将410个差异表达基因利用SVM-RFE算法对MM的诊断基因进行筛选,共得到2个MM诊断基因CPXM1和UROD(图3A);本研究在验证集中进行了CPXM1和UROD基因表达量的校正,均P<0.01(图3B);并绘制ROC曲线,CPXM1和UROD曲线下面积(area under curve,AUC)均为0.947,95%CI=0.842~1.000(图3C),表明筛选出的基因对MM具有较好的诊断价值。
2.4 免疫浸润分析
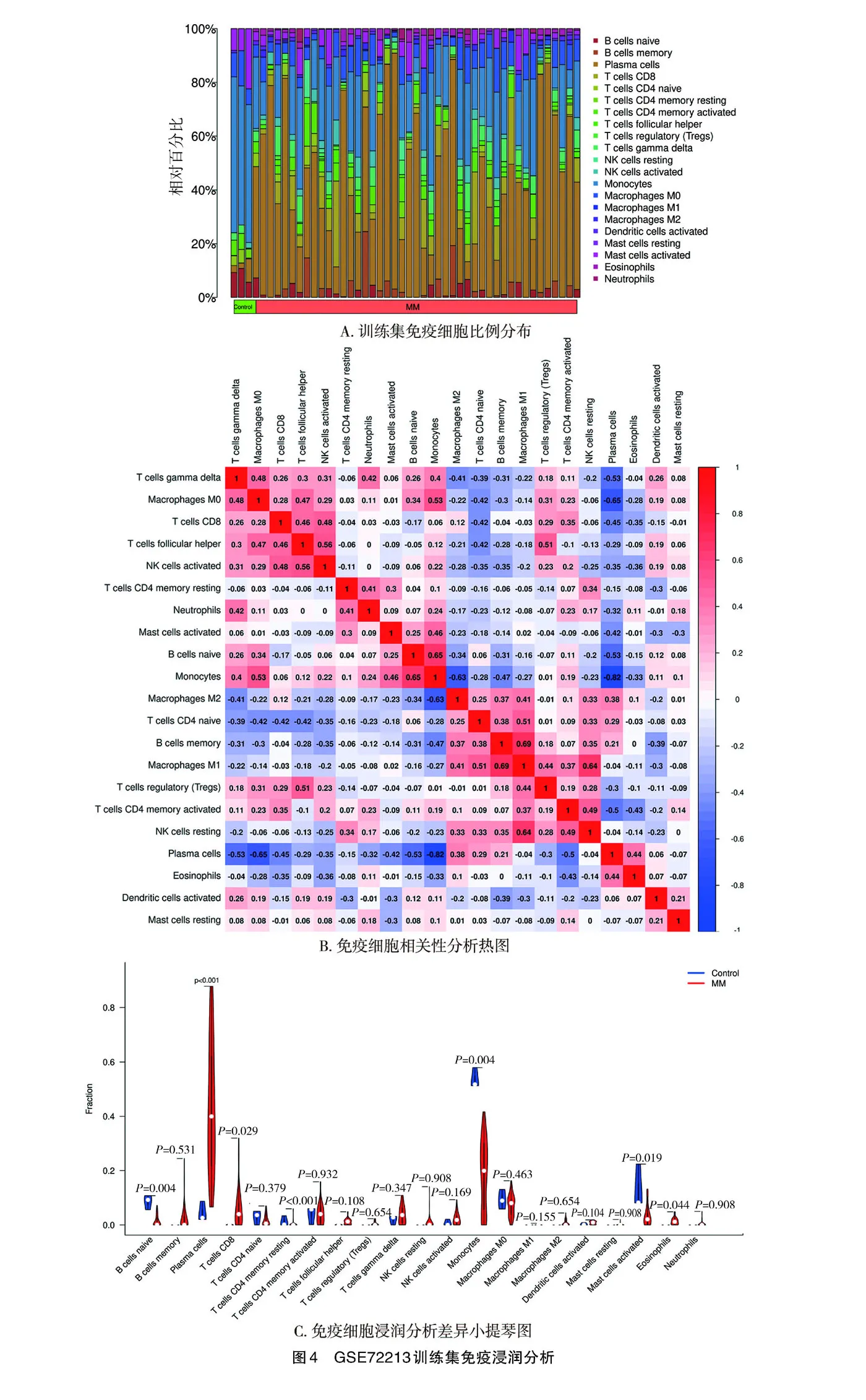
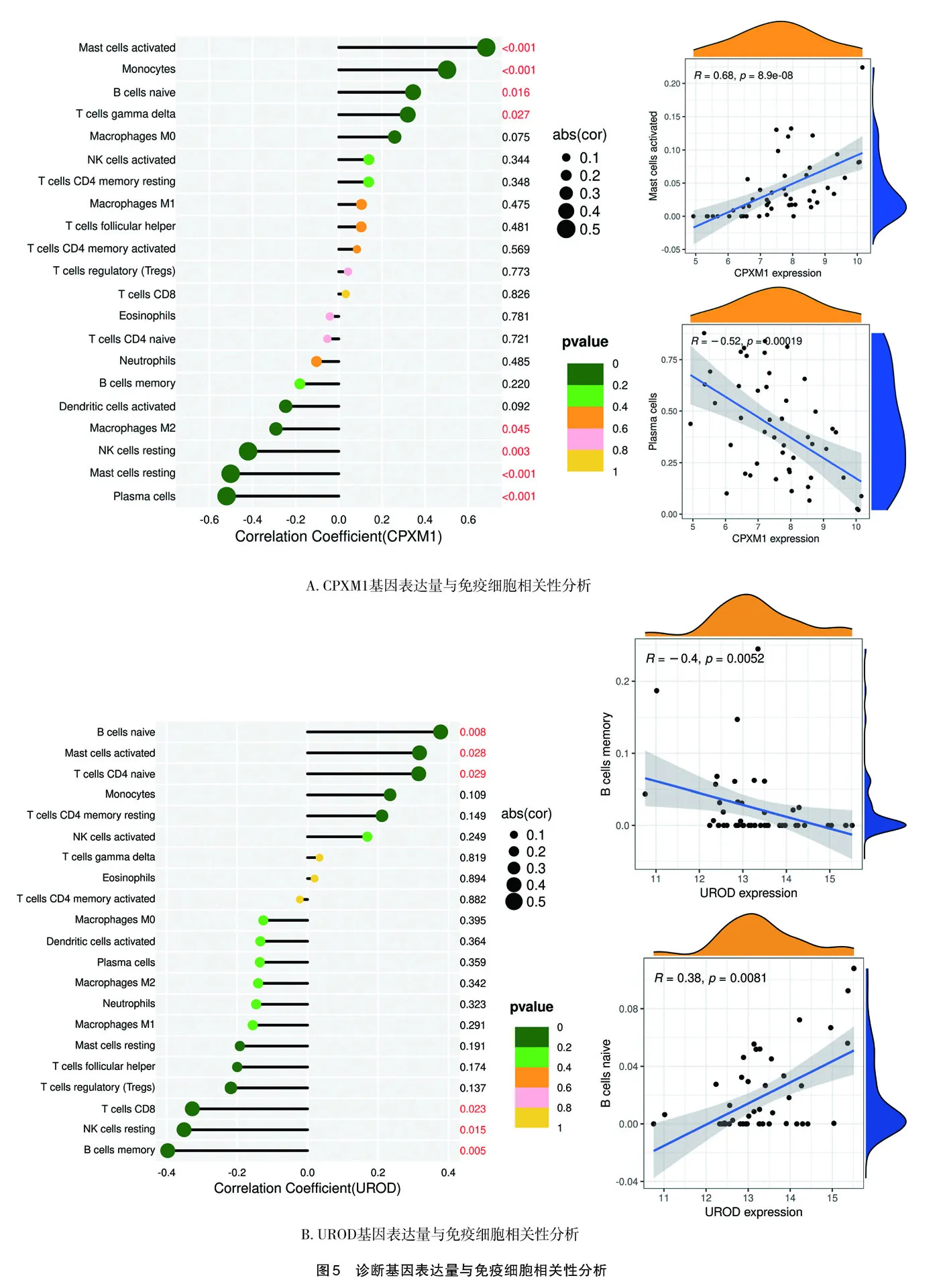
利用CIBERSORTx根据22种免疫细胞的基因表达矩阵,用R语言软件分析GSE72213训练集中MM组和对照组的免疫细胞浸润情况发现两组免疫细胞比例具有明显差异(图4A);同时M1巨噬细胞和记忆性B淋巴细胞显著正相关,浆细胞和单核细胞显著负相关(图4B);MM患者有7类免疫细胞较健康对照组具有统计学差异,分别是幼稚性B淋巴细胞(P=0.004)、浆细胞(P<0.001)、CD8 T淋巴细胞(P= 0.029)、静息期CD4记忆T淋巴细胞(P<0.001)、单核细胞(P=0.004)、活化的肥大细胞(P=0.019)及嗜酸性粒细胞(P= 0.044)(图4C);诊断基因CPXM1表达与免疫细胞相关性分析(图5A)发现,其与浆细胞表达呈负相关(r=-0.520,P= 0.000 19),与活化后的肥大细胞呈正相关(r=0.680,P=8.9×10-8);诊断基因UROD表达与免疫细胞相关性分析(图5B)发现,其与记忆性B淋巴细胞表达呈显著负相关(r=-0.400,P=0.005),与活化后的幼稚性B淋巴细胞呈显著正相关(r= 0.380,P=0.038)。
2.5 诊断基因
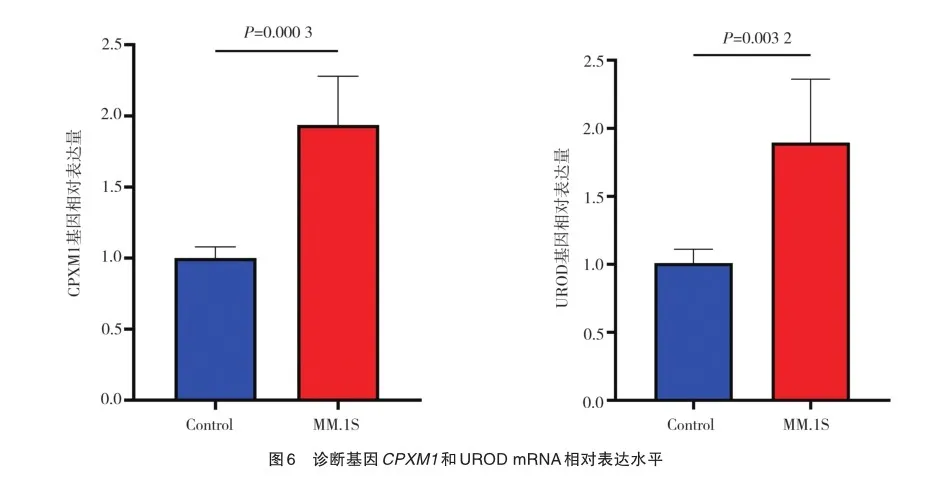
CPXM1和UROD相对表达水平:通过采用免疫磁珠分选正常骨髓CD138阳性浆细胞为对照细胞,将MM.1S细胞为实验组,进行qRT-PCR检测(图6),结果显示MM.1S细胞株CPXM1的相对表达水平为1.936±0.343,与对照细胞(0.999±0.079)比较,差异有统计学意义(t=5.948,P<0.001);UROD的相对表达水平为1.896±0.466,与对照细胞(1.009±0.102)比较,差异有统计学意义(t=4.155,P< 0.010)。
3 讨论
MM是一种恶性血液系统肿瘤,目前治疗以分子药物化疗为主,近年来随着新型治疗方案(免疫调节药物及单克隆抗体等)的临床应用,MM总生存率较以往提高,但患者预后及难治型和复发型患者治疗效果仍差。MM的病因复杂,存在多种诱发因素(辐射、遗传及染色体基因突变等),而骨髓穿刺是临床诊断MM的主要手段,但MM患者具异质性,其瘤细胞可以在髓内呈斑片状浸润(不均匀分布),还可同时或仅以髓外浸润作为临床表现[6]。因此,探究MM的遗传异质性发现早期变化且特异性高的诊断标志物将有助于对患者尽早开展治疗。
本研究对GEO数据库中的基因表达芯片数据文件GSE125364和GSE72213中MM患者和健康对照组的骨髓样本进行了生物信息学数据分析,最终筛选出410个差异基因,MM患者的样本中较健康对照组表达上调的基因主要有LAMP3、CAMK2N2、SCAMP5、DUSP5、HLA-DOB、SDC1、PLA2G16、PERP、PRDM1、MOXD1、HRASLS2、CPNE5、BMP6、GPRC5D及TNFRSF17等,表达下调的基因主要有BEX1、VPREB1、DEFB1、CCL18、TFRC、DNTT、SPTA1、IGLL1、HDC、GFI1B、KLF1、CPA3、C17orf99、AKR1C3及CD36等。
通过GO、KEGG和GSEA分析发现DEGs主要涉及染色体异常、DNA 复制、细胞周期、p53信号通路、脂肪细胞因子信号通路及细胞衰老等方面;而在多发性骨髓瘤(MM)中,导致克隆异质性的主要因素是基因组不稳定性,其使得细胞在分裂和分化过程中更容易产生新的遗传变化,并因此演变成不同的克隆亚群[7];这种不稳定性通常表现为染色体异常和高频突变,绝大多数MM患者都会出现染色体拷贝数的变异[8-9],这种遗传学异常是MM进展和恶化的关键驱动力,影响了患者的诊断和预后。细胞周期蛋白D基因似乎都会在MM早期调节异常,促进细胞周期进展和肿瘤增殖[10]。多种机制都可导致细胞周期蛋白D表达异常,包括涉及CCND1(11q23)、CCND2(12p13)和CCND3(6p21)基因的易位,转录因子MAF诱导的CCND2转录上调,以及CCND1基因点突变[11]。肿瘤抑制蛋白p53表达和功能异常会影响MM的发生和发展,并与患者预后相关[12],此外p53的缺失增加了克隆性MM细胞的肿瘤启动潜力和耐药性[13]。虽然p53在MM中的具体作用机制尚不完全明确,但其功能异常与细胞衰老和凋亡相关。细胞衰老阻止细胞分裂并增加细胞因子的表达和分泌,是抑制肿瘤的重要机制[14]。化疗药物通过诱导凋亡和衰老来抑制肿瘤[15]。衰老和肥胖的MM患者,其骨髓脂肪组织增加,而MM细胞可以改变骨髓脂肪组织的基因表达和细胞因子分泌,影响能量代谢和诱导衰老表型[16];多发性骨髓瘤骨病(multiple myeloma bone disease,MBD)是MM的显著特征,绝大部分的MM患者在首次就诊时就伴随溶骨性病变。Liu H等[17]研究发现,MBD的发生与骨髓脂肪细胞息息相关。MM主要影响中老年人,发病率随年龄增加而上升。根据美国政府官方网站(https://seer. cancer. gov/statfacts/html/mulmy. html)显示,2017年至2021年美国人群中MM的中位发病年龄为69岁,其中65~74岁占比32.2%,45岁以下占比仅为3.1%。
本研究通过机器学习的方法筛选出了MM患者的诊断基因CPXM1和UROD并在验证数据集GSE72213和MM细胞系中进行了验证。羧肽酶(Carboxypeptidases,CPs)在人体生理的各个方面都发挥着至关重要的作用[18]。例如,羧肽酶A3是一种位于肥大细胞中的特殊羧肽酶,参与先天免疫、血管生成和细胞外基质重塑。羧肽酶X成员1(car‐boxypeptidase X member 1,CPXM1)属于CPs家族。有报道称,CPXM1是一种分泌性胶原结合糖蛋白,通过含有160个氨基酸的盘状蛋白结构域与其他蛋白质结合[19]。还已证实CPXM1能够调节脂肪生成并作用于FGF-1/BAMBI的下游,并可能通过影响细胞外基质重塑来促进增生性脂肪组织的扩增[20]。CPXM1对包括恶性肿瘤在内的人体生理生化系统有着显著影响,多项研究报告称,CPXM1可作为卵巢癌[21],乳腺癌[22],颈部鳞状细胞癌[23],骨髓增生异常综合征[24]和乳头状甲状腺[25]的生物标志物,且作为黑色素瘤的新免疫治疗靶点,已证明CPXM1可用作预测抗PD-1治疗结果的生物标志物[26]。本研究发现MM患者CPXM1表达水平较高,然而,CPXM1在MM患者病理生理过程中发挥什么作用尚不清楚,可能是下一步基础研究的重点。
尿卟啉原脱羧酶(Uroporphyrinogen Decarboxyl‐ase,UROD)是血红素生物合成的关键酶,其在肿瘤研究领域中报道较为少见。通过基于RNA干扰的高通量测序UROD被鉴定为头颈部肿瘤选择性放射增敏靶点。UROD敲除加上辐射可在体外诱导头颈部肿瘤细胞中caspase介导的细胞凋亡和细胞周期停滞,并抑制头颈部肿瘤细胞体内的肿瘤形成能力,延迟小鼠体内已建立的肿瘤异种移植物的生长。此外,UROD在头颈部肿瘤患者活检中显着过表达。较低的放射前UROD mRNA表达与改善的无病生存率相关,UROD下调还使不同的癌症模型(肺癌、宫颈癌、前列腺癌和乳腺癌)放疗敏感,并使肿瘤对化疗药物(包括5-氟尿嘧啶、顺铂和紫杉醇)敏感[27]。本研究发现MM患者UROD表达水平较高,进一步探究UROD在MM中的分子机制可能有助于研发MM患者新的治疗方案。
本研究免疫浸润分析显示,浆细胞、CD8 T淋巴细胞和嗜酸性粒细胞是多发性骨髓瘤患者较健康人群升高的免疫浸润细胞,幼稚性B淋巴细胞、静息期CD4记忆T淋巴细胞、单核细胞和活化的肥大细胞是较健康人群降低的免疫浸润细胞,且M1型巨噬细胞和记忆性B淋巴细胞、单核细胞和幼稚性B淋巴细胞及单核细胞和浆细胞相关性显著;MM的主要效应细胞是破骨细胞,它们来源于单核-巨噬细胞系。MM细胞和骨髓微环境中的其他细胞刺激这些破骨细胞,导致其增殖并变得功能活跃,进而引发溶骨亢进。这与MM的主要效应细胞的起源一致,破骨细胞在MM疾病的发生和发展中起到关键作用。此外本研究使用CIBERSORTx探究诊断基因CPXM1和UROD的表达与免疫细胞浸润之间的关系发现CPXM1基因的表达与活化的肥大细胞、单核细胞及幼稚性B淋巴细胞呈正相关,与浆细胞、静息的肥大细胞及静息的NK细胞呈负相关,同时UROD的表达与幼稚性B淋巴细胞、活化的肥大细胞及幼稚性CD4 T淋巴细胞呈正相关,与记忆性B淋巴细胞、静息的NK细胞及CD8 T淋巴细胞呈负相关,而既往研究表明,免疫微环境中效应细胞比例的升高往往提示着患者的较好预后。这些结果帮助更好地理解CPXM1和UROD在多发性骨髓瘤中的作用及其对免疫微环境的影响。
综上所述,本研究基于公共数据库的多发性骨髓瘤基因芯片数据采用生物信息学方法分析筛选出多发性骨髓瘤2个诊断基因,并表明诊断基因与免疫浸润相关,但仍有一些局限性,本研究所纳入的研究数据样本数量有限,样本类型仅选择了骨髓样本,存在选择偏倚;由于MM患者的异质性,本课题组所筛选的诊断基因对具有不同遗传异质性的MM患者是否适用需要相关实验数据证实;且CPXM1和UROD在MM患者疾病发生发展过程中具体参与的分子机制仍需进一步探究。
参考文献
[1] Cowan AJ,Green DJ,Kwok M,et al. Diagnosis and management of multiple myeloma:a review[J]. JAMA,2022,327(5):464-477.
[2] Bazarbachi AH,Al Hamed R,Malard F,et al. Relapsed refractory multiple myeloma:a comprehensive overview[J]. Leukemia,2019,33(10):2343-2357.
[3] Kumar SK,Rajkumar SV,Dispenzieri A,et al. Improved survival in multiple myeloma and the impact of novel therapies[J]. Blood,2008,111(5):2516-2520.
[4] Moreau P,Kumar SK,Miguel JS,et al. Treatment of relapsed and refractory multiple myeloma:recommendations from the International Myeloma Working Group[J]. Lancet Oncol,2021,22(3):e105-e118.
[5] Arron JR,Choi Y. Bone versus immune system[J]. Nature,2000,408(6812):535-536.
[6] Huang BH,Li J. Advances in the diagnosis and treatment of mul‐tiple myeloma[J]. Zhonghua Xue Ye Xue Za Zhi,2018,39(7):605-608.
[7] Bakhoum SF,Landau DA. Chromosomal instability as a driver of tumor heterogeneity and evolution[J]. Cold Spring Harb Perspect Med,2017,7(6):a029611.
[8] Neuse CJ,Lomas OC,Schliemann C,et al. Genome instability in multiple myeloma[J]. Leukemia,2020,34(11):2887-2897.
[9] Manier S,Salem KZ,Park J,et al. Genomic complexity of multiple myeloma and its clinical implications[J]. Nat Rev Clin Oncol,2017,14(2):100-113.
[10] Bergsagel PL,Kuehl WM,Zhan FH,et al. Cyclin D dysregula‐tion:an early and unifying pathogenic event in multiple myeloma[J]. Blood,2005,106(1):296-303.
[11] Maura F,Petljak M,Lionetti M,et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines[J]. Leukemia,2018,32(4):1043-1047.
[12] Walker BA,Mavrommatis K,Wardell CP,et al. A high-risk,Double-Hit,group of newly diagnosed myeloma identified by genomic analysis[J]. Leukemia,2019,33(1):159-170.
[13] Chang YT,Chiu I,Wang QJ,et al. Loss of p53 enhances the tumor-initiating potential and drug resistance of clonogenic multiple my‐eloma cells[J]. Blood Adv,2023,7(14):3551-3560.
[14] Kuilman T,Peeper DS. Senescence-messaging secretome:SMSing cellular stress[J]. Nat Rev Cancer,2009,9(2):81-94.
[15] Acosta JC,Gil J. Senescence:a new weapon for cancer therapy[J]. Trends Cell Biol,2012,22(4):211-219.
[16] Fairfield H,Dudakovic A,Khatib CM,et al. Myeloma-modified adipocytes exhibit metabolic dysfunction and a senescence-associated secretory phenotype[J]. Cancer Res,2021,81(3):634-647.
[17] Liu H,He J,Koh SP,et al. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease[J]. Sci Transl Med,2019,11(494):eaau9087.
[18] Sapio MR,Fricker LD. Carboxypeptidases in disease:insights from peptidomic studies[J]. Proteomics Clin Appl,2014,8(5/6):327-337.
[19] Kim YH,O’Neill HM,Whitehead JP. Carboxypeptidase X-1(CPX-1) is a secreted collagen-binding glycoprotein[J]. Biochem Bio‐phys Res Commun,2015,468(4):894-899.
[20] Kim YH,Barclay JL,He JJ,et al. Identification of carboxypepti‐dase X(CPX)-1 as a positive regulator of adipogenesis[J]. FASEB J,2016,30(7):2528-2540.
[21] Zheng MJ,Long JY,Chelariu-Raicu A,et al. Identification of a novel tumor microenvironment prognostic signature for advanced-stage serous ovarian cancer[J]. Cancers,2021,13(13):3343.
[22] Mao XH,Ye Q,Zhang GB,et al. Identification of differentially methylated genes as diagnostic and prognostic biomarkers of breast can‐cer[J]. World J Surg Oncol,2021,19(1):29.
[23] Chen Y,Li ZY,Zhou GQ,et al. An immune-related gene prognos‐tic index for head and neck squamous cell carcinoma[J]. Clin Cancer Res,2021,27(1):330-341.
[24] Wang YH,Lin CC,Yao CY,et al. A 4-gene leukemic stem cell score can independently predict the prognosis of myelodysplastic syn‐drome patients[J]. Blood Adv,2020,4(4):644-654.
[25] Kumar A,Bandapalli OR,Paramasivam N,et al. Familial Cancer Variant Prioritization Pipeline version 2(FCVPPv2) applied to a papil‐lary thyroid cancer family[J]. Sci Rep,2018,8:11635.
[26] Tian LY,Long F,Hao YJ,et al. A cancer associated fibroblastsrelated six-gene panel for anti-PD-1 therapy in melanoma driven by weighted correlation network analysis and supervised machine learning[J]. Front Med,2022,9:880326.
[27] Ito E,Yue SJ,Moriyama EH,et al. Uroporphyrinogen decarboxyl‐ase is a radiosensitizing target for head and neck cancer[J]. Sci Transl Med,2011,3(67):e3001922.
(责任编辑:李青颖)
本文引用格式:
石 蕾,张红宾. 基于SVM机器学习筛选多发性骨髓瘤的特征基因及免疫浸润分析[J]. 重庆医科大学学报,2025,50(1):135-144.
猜你喜欢
中国中药杂志(2016年21期)2017-02-16 14:10:12
中国中药杂志(2016年21期)2017-02-16 13:04:42
云南中医中药杂志(2016年11期)2016-12-26 17:40:37
中国教育信息化·基础教育(2016年10期)2016-12-20 20:09:27
中国当代医药(2016年28期)2016-12-20 18:16:18
今传媒(2016年11期)2016-12-19 11:35:50
电脑知识与技术(2016年27期)2016-12-15 19:19:25
中国科技纵横(2016年17期)2016-11-30 21:40:27
中国实用医药(2016年17期)2016-07-26 00:33:52
中国实用医药(2016年13期)2016-07-05 13:43:48
