
基于代谢组学研究TetR家族转录因子SPA7074对密旋链霉菌Act12代谢的影响
2024-12-31 00:00:00郝鹏泽赵康康张媛陈欢薛泉宏贾良辉颜华
西北农业学报 2024年11期


摘 要" 为了阐明TetR家族转录因子SPA7074影响密旋链霉菌Act12的具体机制,利用超高效液相色谱-质谱联用的非靶向代谢组学方法,检测了SPA7074缺失突变株(Δspa7074)与野生型Act12之间的胞内代谢物图谱,并进行多元筛选统计分析。结果表明:在正离子模式下Δspa7074中相较于野生型显著差异代谢物共有55种,为氨基酸及其衍生物、核黄素、S腺苷甲硫氨酸和低碳有机酸等物质,这些化合物多为寡霉素等大环内酯类抗生素生物合成的前体或中间产物,以及能量代谢相关物质;通过胞内相关的次级代谢中间产物的结构分析,对寡霉素的生物合成途径进行了初步的阐述。以上结果说明SPA7074敲除后胞内寡霉素生物的合成显著提高并及时转运出胞外,消耗了大量的氨基酸代谢相关产物,增加了细胞的能量供应,改变了胞内的代谢流。
关键词 密旋链霉菌;非靶向代谢组学;寡霉素D
密旋链霉菌Act12(Streptomyces pactum)是西北农林科技大学薛泉宏教授课题组分离得到的一株生物防治菌,其能抑制真菌病原体、促进植物生长[1-2]、改变植物根际微生物群落[3]、促进植物次生代谢产物积累[4]。
TetR家族是一类在原核生物中广泛分布的转录因子,由保守的螺旋-转角-螺旋DNA结合结构域和C端的配体调节结构域组合而成。其可调节微生物渗透压及生理平衡、促进抗生素的生物合成、影响不同代谢通路中蛋白的活性。前期研究发现,TetR家族转录因子SPA7074敲除突变株Δspa7074可显著提高多种植物对病原真菌的拮抗能力,HPLC结果表明,与Act12野生型相比,Δspa7074中多种代谢物质发生了较大改变,特别是抑菌物质寡霉素D的产量最高,是野生型的7倍多[5]。
作为一种新兴组学工具,代谢组学借由高效液相色谱[6]、核磁共振波谱[7]和质谱技术的发展,广泛应用于食品科学[8-9]、植物学[10-11]以及医学研究[12-13],与转录组、蛋白组学一起成为研究生物体代谢表达活动的重要手段之一[14]。然而,目前借助代谢组手段对微生物的研究更多应用于植物微生态[15]和人体微生态[16]等方面,针对单一菌株突变体代谢组变化的研究相对较少。为此,本研究利用基于超高效液相色谱-质谱联用的非靶向代谢组学方法[17-18],分析了TetR家族转录因子SPA7074敲除突变株(Δspa7074)与Act12野生型之间整体差异代谢物,并利用层次聚类分析、差异代谢物相关分析、KEGG富集分析等方法,筛选了突变株与野生型之间的差异代谢通路,可为揭示寡霉素的合成途径奠定基础。
1 材料与方法
1.1 主要材料
密旋链霉菌Act12(Streptomyces pactum)野生型(WT)、SPA7074敲除突变株(Δspa7074),均来源于西北农林科技大学放线菌分子生物学实验室保藏菌株;TSB培养基:大豆蛋白胨3 g/L,胰蛋白胨17 g/L,NaCl 5 g/L,K2HPO4 2.5 g/L,葡萄糖2.5 g/L,121 ℃灭菌25 min。
1.2 主要试剂
10×binding buffer,50%甘油,0.1" mol/L MgCl2,1% NP40,20 mmol/L EDTA,10×TBE,10%过硫酸铵(APS),均由本实验室配置;SYBR safe DNA凝胶染料,购自thermo fisher;6×DNA loading buffer,购自擎科生物。
1.3 胞内代谢物的制备和提取
将活化的密旋链霉菌Act12野生型(WT),SPA7074敲除突变株(Δspa7074)接种到TSB培养基中,28 ℃、150 r/min培养9 d后取样,3种菌株样本设置6个重复。4 ℃、5 000 r/min离心 "5 min收集菌体3次,使用预冷的PBS悬浮菌体;弃去全部上清液,将菌体分装为0.1 mL收集在离心管中,液氮速冻,-80 ℃保存。
取一管分装后的菌体样本,加入500 μL含0.1%甲酸的80%甲醇溶液(均为质量分数),涡旋混匀,冰浴静置5 min,4 ℃、15 000 r/min离心10 min,收集上清液到新离心管中,加入150 μL超纯水,涡旋混匀后,转移到带有0.22 μm滤膜的离心管中,4 ℃、15 000 r/min离心10 min,收集滤液到进样瓶进行质谱联用分析。
1.4 Δspa7074与野生型间差异代谢物筛选
在液质联用测定后,先将得到的代谢组数据进行图谱匹配和搜库,拟合分析相应的代谢物种类及含量;再进行质控以保证数据的准确性,筛选统计相关数据。通过变量投影重要度(VIP值)和每个代谢物在2个处理中所有生物重复定量值的均值之比(FC值),结合t检验进行筛选,并绘制展示比较对(WT vs Δspa7074)间的差异代谢物整体分布火山图。
1.5 Δspa7074与野生型间代谢物差异分析
1.5.1 差异代谢物相关性分析 计算比较对WT vs Δspa7074间差异代谢物间皮尔逊相关系数,进行代谢物相关性分析,并进行显著性统计检验[19],Plt;0.05即为显著性差异。
1.5.2 差异代谢物功能分析 KEGG(Kyoto Encyclopedia of Genes and Genomes)是进行生物体内代谢分析、代谢网络研究的重要方法[20-23]。对鉴定到的代谢物利用KEGG进行功能和分类注释,并在KEGG富集图的基础上进一步解析代谢物所参与的生化途径以及信号转导路线,使用超几何检验计算阈值(P-value),进一步分类找出胞内关键代谢物所在通路。
2 结果与分析
2.1 Δspa7074与野生型Act12间的差异代谢物分析
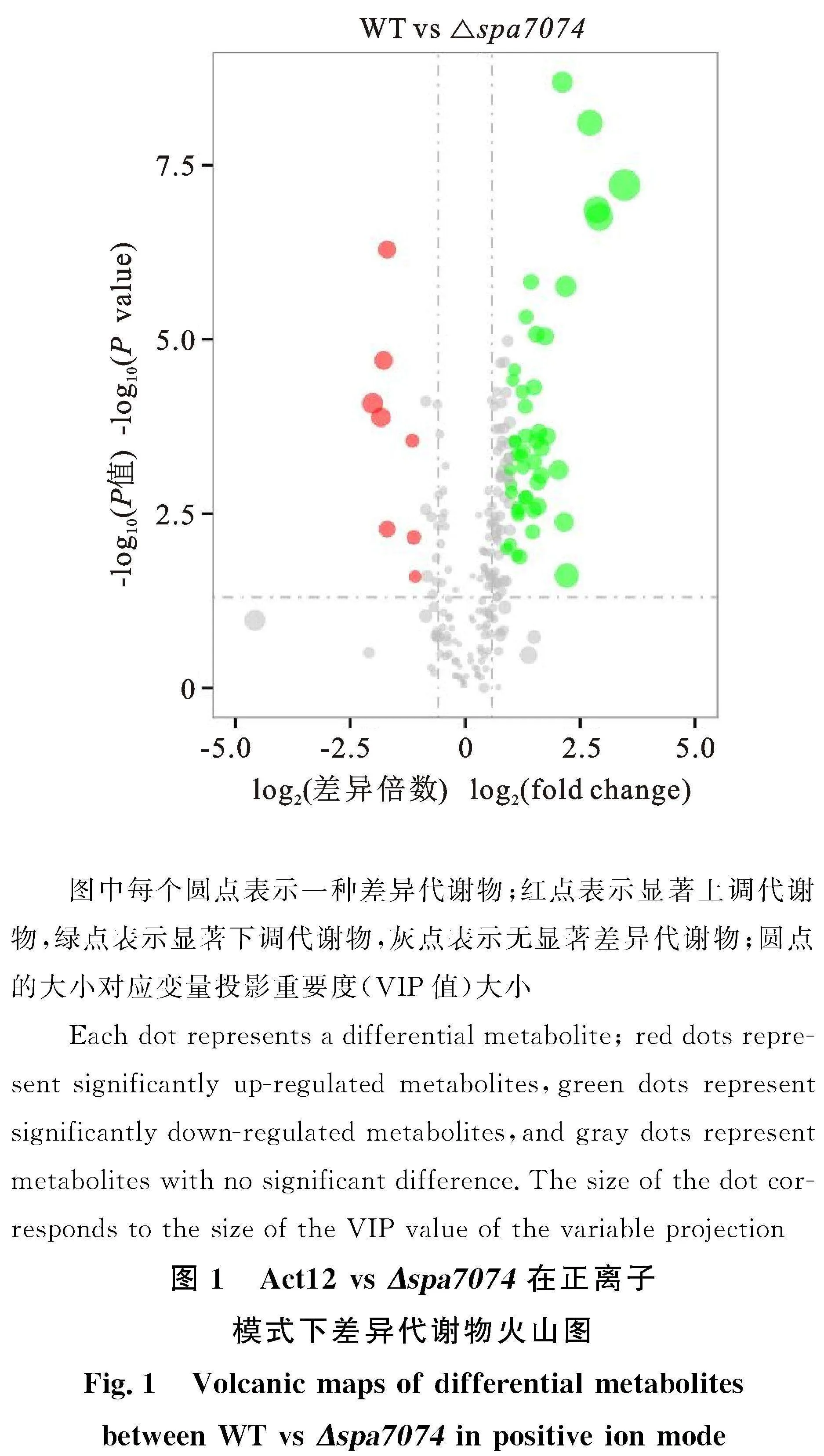
筛选VIPgt;1且Plt;0.05的代谢物,视为差异代谢物。由图1可知,在正离子模式下,Δspa7074与Act12间的差异代谢物共有55种,其中47种显著下调,8种显著上调。表1列出了其中差异最显著的10种化合物。
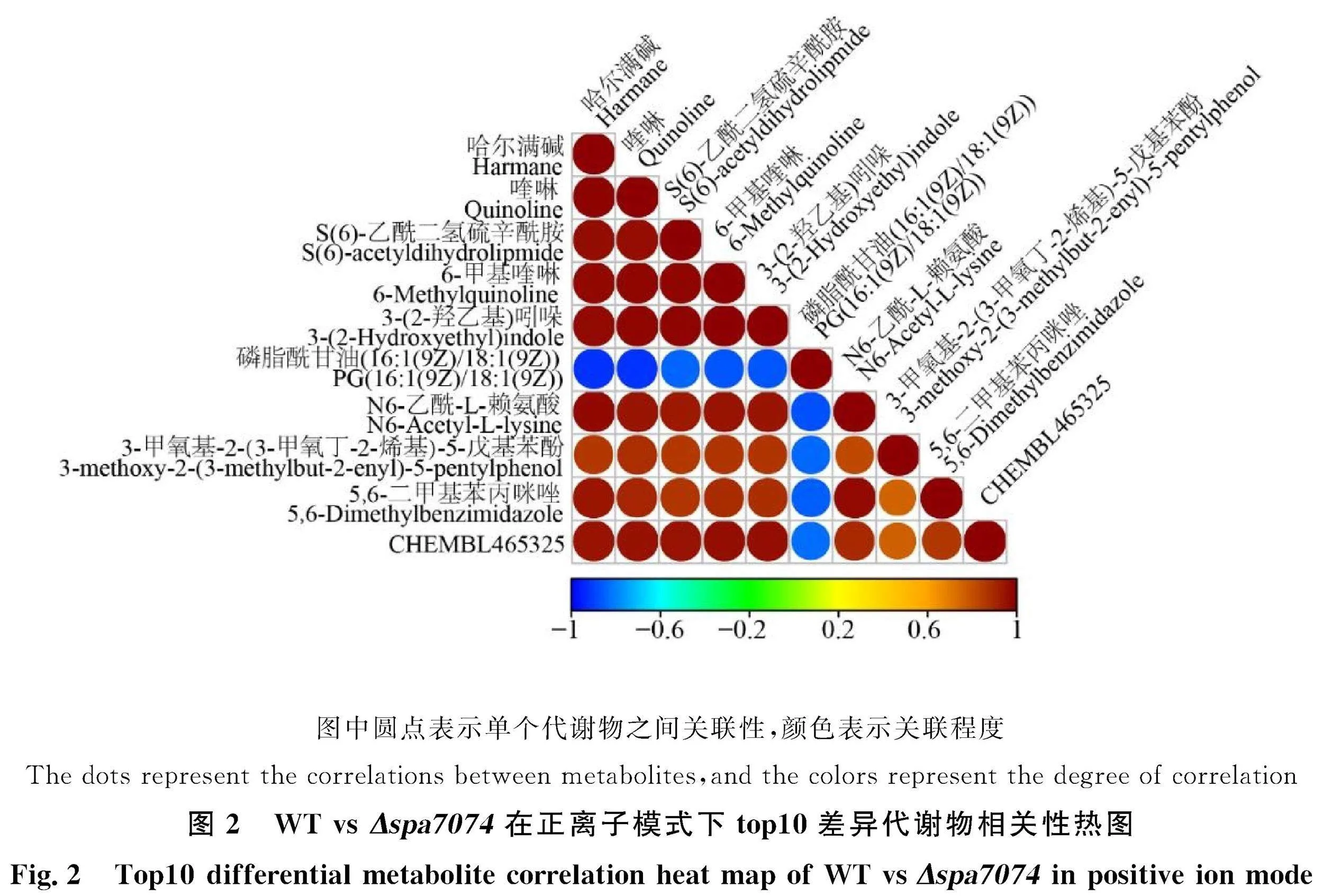
2.2 Δspa7074与野生型Act12间代谢物相关性分析
为进一步探究代谢物之间的关联性,对得到的差异代谢物进行相关性分析(图2)。结果显示,上调的8种代谢物差异代谢物具有强正相关性;下调的47种差异代谢物间也具有正相关性,且上、下调的差异代谢物两两间具有较强的负相关性。另外,显著上调的差异代谢物主要是次生代谢产物及其前体化合物,而显著下调的代谢物主要是初生代谢产物与细胞能量代谢相关化合物。因此,初步推断在敲除 "SPA7074基因后,胞内初生代谢产物与能量代谢产物消耗速度加快,转化为次生代谢产物,并使其积累量增加。
2.3 KEGG富集分析
在正离子模式下分析差异代谢物的富集通路,发现次生代谢产物种类较多,但大部分未能富集在通路上(图3)。在上调的差异代谢物中有6种未能被富集在通路中,包括:磷脂酰甘油(16:1(9Z)/18:1(9Z))、15-脱氧-Δ12,14-前列腺素A1、松脂多辛、人参皂苷 Rg2、1-硬脂酰基-2-油酰基-sn-甘油、2,3-二甲氧基-5-甲基-6-(3-甲基-2-丁烯-1-基)-1,4-苯二醇;其他上调差异代谢产物富集在C5支化二元酸代谢与丙酸盐代谢通路中;在下调的差异代谢物中有10种差异代谢化合物未能富集在通路中,包括:3-甲氧基-2-(3-甲基丁-2-烯基)-5-戊基苯酚、CHEMBL465325、2-花生四烯酰甘油、3-甲氧基-2-(3-甲基丁-2-烯基)-5-戊基苯酚、19(R)-HETE、5′-氨基甲酰胺-5′-脱氧腺苷、脯氨酰亮氨酸、L-α-亚麻酸-γ-D-Glu-内消旋二氨基庚二酸、N(2)-琥珀酰-L-鸟氨酸和2-庚基-4-羟基喹啉n-氧化物。除却未被富集的差异代谢物,被富集的通路大多为次生产物、各类氨基酸以及能量的代谢通路。与野生型相比,突变株中氨基酸、部分次生代谢产物、能量相关化合物相关中间体的合成与代谢表达量均有明显变化。
3 讨" 论
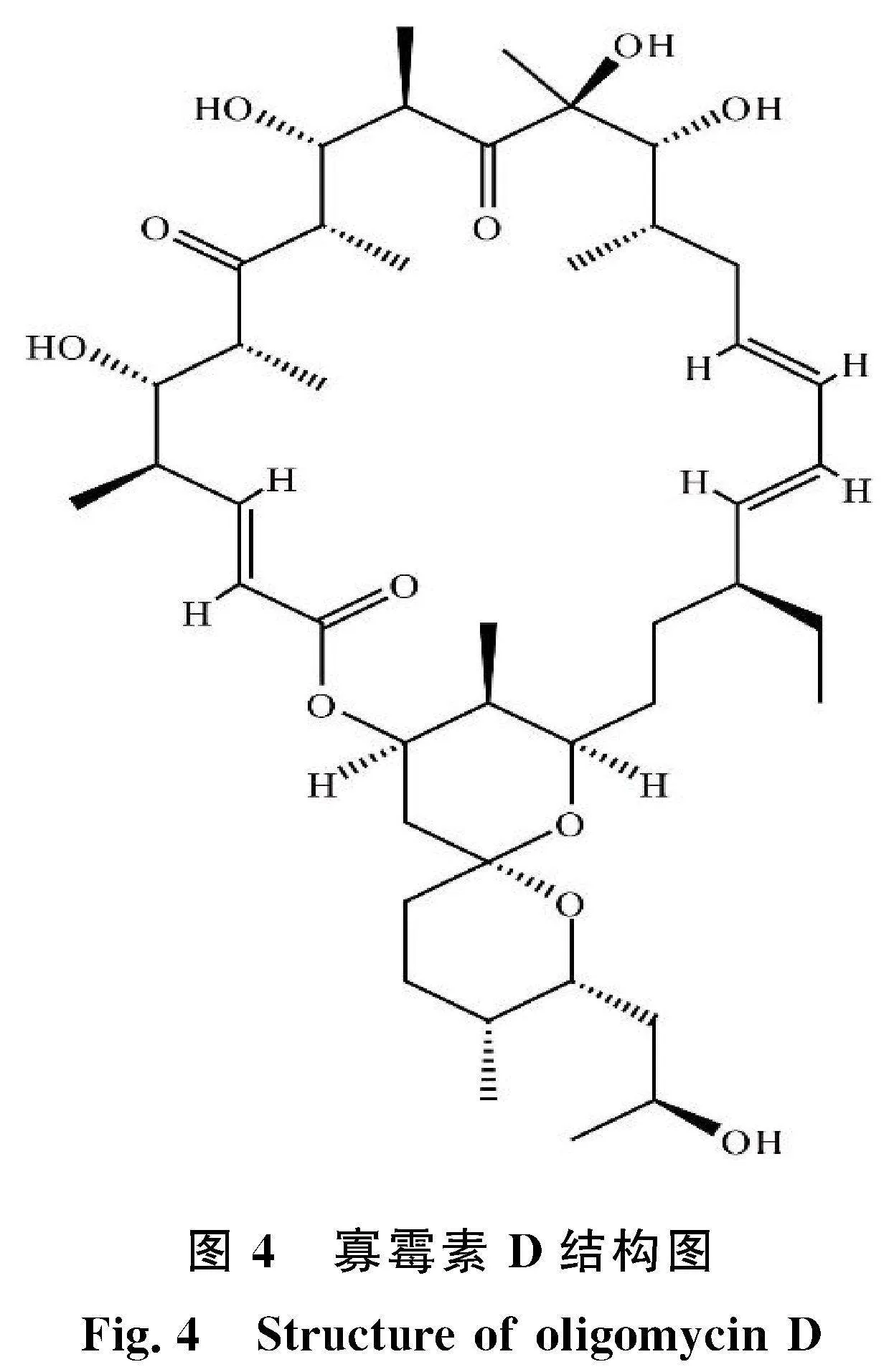
链霉菌Act12野生型的基因簇大部分都是沉默或低表达的,前期高效液相色谱检测结果显示[5],敲除SPA7074后突变株产生的寡霉素D的含量与野生型相比上升了7倍(图4)。而通过转录组分析发现寡霉素生物合成基因簇内多个基因表达量提高,与SPA7074在染色体上相邻的通道蛋白RS00415的转录水平也提高了10倍左右[24],因此寡霉素D生物合成量的增加可能与SPA7074敲除后,解除了对这些基因的转录抑制有关,寡霉素生物合成基因簇内相关合成基因的表达水平可以提高寡霉素的合成量,通道蛋白表达水平的提高可以及时将寡霉素转运出胞外,从而解除寡霉素生物合成的产物反馈抑制。
而在本研究中,相较于野生型,突变株Δspa7074中氨基酸代谢产物、部分次生代谢物质、能量代谢中间体表达量下降。这可能是寡霉素生物合成的显著提高消耗了这些化合物,因为氨基酸代谢相关产物可作为寡霉素等聚酮类化合物的合成原料,寡霉素的大量合成也需要细胞更的供给能量;而部分代谢物质的产量下降则一方面可能也是作为寡霉素的合成原料被消耗,也可能是由于寡霉素的合成增加消耗了这些共同前体化合物,从而降低了其他次级代谢产物的合 "成量。
另外,研究者前期研究表明,寡霉素D由1,7-二氧杂螺[5.5]十一烷环的螺缩酮加一个十七碳的聚酮的结构组成[25],而此聚酮结构的形成只需要活化的乙酸盐,甲基丙二酰或者其活化的形式(丙二酰辅酶A),活化的丁酸盐就可以不断缩合,形成不断增长的聚酮类化合物链[26],而在此缩合过程中,只需要十一碳大小的二羟基酮即可合成所需的1,7-二氧杂螺[5.5]的螺缩酮[27]。因此推测在此过程中,多种氨基酸产物,部分次生代谢物质、能量代谢中间体的合成可能与此有一定联系,且其中8种显著上调的差异代谢物可能与寡霉素D合成有较强相关性。然而目前寡霉素D的生物合成通路并未阐明,且多数次生代谢产物合成通路也缺乏完整报道,目前还无法准确揭示显著上调代谢物影响寡霉素D合成的相关 "机制。
本试验通过超高相液相色谱-质谱联用的非靶向代谢组学方法,研究发现链霉菌Act12在在敲除转录因子SPA7074后,其代谢产物与野生型相比发生显著变化,其中8种显著上调,47种显著下调,且上、下调差异代谢物之间有较强的负相关性。上调富集通路主要集中在C5支化二元酸通路、丙酸盐代谢通路两个通路中,他们可能与寡霉素D合成存在联系。本研究可为揭示寡霉素D的生物合成途径奠定基础,为生防链霉菌Act12在农业生产中的应用提供理论参考。
参考文献 Reference:
[1] 申光辉,薛泉宏,陈 秦,等.硅酸钾与密旋链霉菌Act12菌剂配施对连作草莓生长、果实产量及品质的影响[J].中国生态农业学报,2012,20(3):315-321.
SHEN G H,XUE Q H,CHEN" Q,et al.Effects of combined application of potassium silicate and Streptomyces pactum bio-control agents on growth,yield and quality of strawberry under continuous cropping in greenhouse [J].Chinese Journal of Eco-Agriculture,2012,20(3):315-321.
[2]周金华,杨晶晶,惠 珂,等.3株放线菌对白菜根肿病的防治效果及机制研究[J].西北农业学报,2020,29(4):641-651.
ZHOU J H,YANG J" J,HUI K,et al.Effects of 3 biocontrol strains on clubroot disease-resistant and" mechanism in Chinese cabbage [J].Acta Agriculturae Boreali-occidentalis Sinica,2020,29(4):641-651.
[3]LI Y,GUO Q,LI Y,et al.Streptomyces pactum Act12 controls tomato yellow leaf curl virus disease and alters rhizosphere microbial communities[J].Biology and" Fertility of" Soils,2019,55(2):149-169.
[4]阎 岩,赵 欣,张顺仓,等.活性氧在密旋链霉菌Act12诱导丹参毛状根中丹参酮积累中的作用[J].中国中药杂志,2014,39(11):1985-1991.
YAN Y,ZHAO X,ZHANG SH" C,et al.Roles of reactive oxygen species in Streptomyces" pactum Act12-induced tanshinone production in Salvia miltiorrhiza hairy roots [J].China Journal of Chinese Materia Medica,2014,39(11):1985-1991.
[5]段雪梅,赵飞扬,颜 霞,等.生防菌密旋链霉菌Act12中SPA7074缺失突变株的构建及其次级代谢产物鉴定[J].微生物学报,2016,56(12):1883-1891.
DUAN X M,ZHAO F Y,YAN" X,et al.Construction of SPA7074-deficient mutant of biocontrol strain Streptomyces pactum Act12 and characterization of its secondary metabolites [J].Acta microbiologica Sinica,2016,56(12):1883-1891.
[6]WOLFENDER J,NUZILLARD J,VAN D H J,et al.Accelerating metabolite" identification in natural" product research:toward an ideal combination of" liquid chromatography-high-resolution tandem mass spectrometry and NMR" profiling,in silico databases,and chemometrics[J].Analytical Chemistry,2019,91(1):704-742.
[7]LETERTRE M P M,DERVILLY G,GIRAUDEAU P.Combined nuclear magnetic resonance spectroscopy and" mass spectrometry approaches for metabolomics [J]. "Analytical Chemistry,2021,93(1):500-518.
[8]AUGUSTIN S,LORRAINE B,CLAUDINE M,et al.The food metabolome:a window over dietary exposure [J].The American Journal of Clinical Nutrition,2014,99(6):1286-1308.
[9]GONZALEZ-UARQUIN F,KENZ ,RODEHUTSCORD M,et al.Dietary phytase and myo-inositol supplementation are associated with distinct plasma metabolome profile in broiler chickens [J].Animal,2020,14(3):549-559.
[10] WANG J X,ZHANG X Q,SHI M L,et al.Metabolomic analysis of the salt-sensitive mutants reveals changes in" "amino acid and fatty acid composition important to long-term salt stress in Synechocystis sp.PCC 6803 [J].Funct Integr Genomics,2014,14(2):431-440.
[11]FUKUSHIMA A,KUSANO M,MEJIA R F,et al.Metabolomic characterization of knockout mutants in Arabidopsis:development of a metabolite profiling database for knockout" mutants in Arabidopsis[J].Plant Physiolagy,2014,165(3):948-961.
[12]YU Y,TAN P,ZHUANG Z,et al.Untargeted metabolomic approach to study the serum metabolites in women with polycystic ovary syndrome [J].BMC Med Genomics,2021,14(1):206.
[13]NASCENTES M,LESNER NP,SABATIER M,et al. "Emerging metabolomic tools to study cancer metastasis [J].Trends Cancer,2022,8(12):988-1001.
[14]INDHUPRIYA S,SRIKANT V,SHIVA K,et al.Multi-omics data" integration,interpretation,and its application[J].Bioinformatics and Biology Insights,2020,14:1-24.
[15]SNEHA G,MARTINO S,UTE R.Metabolomics as an" "emerging tool to study plant-microbe interactions [J]. "Emerging" Topics in" Life Sciences,2022,6(2):175-183.
[16]KUMAR R,SINGH N,BALAKRISHNAN S,et al.Metabolic modeling of the" international space station microbiome reveals key microbial interactions [J].Microbiome,2022,10(1):102.
[17]DUNN W B,DAVID B,PAUL B,et al.Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry [J].Nature Protocols,2018,67(7):1060-1083.
[18]ELIZABETH J W,IAN D W,HELEN G,et al.Global metabolic profiling procedures for urine using UPLC-MS [J].Nature Protocols,2010,5(6):1005-1018.
[19]RAO G,SUI J,ZHANG J.Metabolomics reveals significant variations in metabolites and correlations regarding the maturation of walnuts (Juglans regia L.) [J].Biology Open,2016,5(6):829-836.
[20]KANEHISA M,GOTO S.KEGG:kyoto encyclopedia of genes and genomes [J].Nucleic Acids" Research,2000,28(1):27-30.
[21]RAO J,CHEN F,HU C,et al.Metabolic map of mature maize kernels [J].Metabolomics,2014,10(5):775-787.
[22]LIN H,RAO J,SHI J,et al.Seed metabolomic study reveals significant metabolite variations and correlations" "among different soybean cultivars [J].Journal of Integrative Plant" Biology,2014,56(9):826-836.
[23]JIA S S,WANG Y,HU J,et al.Mineral and metabolic profiles in tea leaves and flowers during flower development [J].Plant Physiology and Biochemistry,2016,106:316-326.
[24]董萌萌.密旋链霉菌Act12中两个TetR家族转录因子调控寡霉素生物合成的研究[D].陕西杨凌:西北农林科技大学,2020.
DONG M M.The regulation of" TetR family transcription factors on the biosynthesis of oligomycin in Streptomyces pactum "Act12 [D].Yangling" Shannxi:Northwest Aamp;F University,2020.
[25]ZHANG F M,ZHANG S Y,TU Y Q.Recent progress in the isolation,bioactivity,biosynthesis,and total synthesis of natural spiroketals [J].Natural Product Reports,2018,35(1):75-104.
[26]LIANG D,YANG X,LIU J,et al.Global evolution of glycosylated polyene macrolide antibiotic biosynthesis [J].Molecular Phylogenetics and Evolution,2018,127:239-247.
[27]ZHENG Q,TIAN Z,LIU W.Recent advances in understanding the enzymatic reactions of [4+2] cycloaddition and spiroketalization [J].Current Opinion in Chemical Biology,2016,31:95-102.
Impact of TetR Family Transcription Factor SPA7074 on Metabolism of
Streptomyces pactum Act12 Based on Metabolomics Research
Abstract To elucidate the specific mechanism by which the TetR-family transcription factor SPA7074 affects the Streptomyces pactum Act12. a non-targeted metabolomic approach using ultra-high-performance liquid chromatography-tandem mass spectrometry was used to detect the intracellular metabolite profiles between the SPA7074 deletion mutant ( Δspa7074 ) and the wild-type Act12, followed by multivariate screening statistical analysis. Ultimately, 55 metabolites were found to be significantly differentially expressed in "Δspa7074" compared to the wild-type in positive ion mode, including amino acids and their derivatives, riboflavin, S-adenosylmethionine, and low-carbon organic acids, which are mostly precursors or intermediates of oligomycin-like macrolide antibiotics biosynthesis, as well as energy metabolism-related substances. The structural analysis of the intermediate products of secondary metabolites related to intracellular biosynthesis pathway of oligomycin D was conducted, which provided a preliminary description of the biosynthesis pathway of oligomycin D. These results indicate that knocking out of SPA7074 significantly increased the intracellular biosynthesis of oligomycin and timely transported it out of the cell, consuming a large amount of amino acid metabolism-related products, increasing the energy supply of cells, and changing the metabolic flow in cells.
Key words Streptomyces pactum; Non-targeted metabolomics; Oligomycin D
