
油茶sRNA分析及其对光合油脂代谢的调控
2024-11-07 00:00王保明尹迎峰颜廷骥张元华陈永忠
经济林研究 2024年3期





关键词:油茶;sRNA;miRNA;靶基因;调控;光合油脂代谢
中图分类号:S601;S794.4 文献标志码:A 文章编号:1003—8981(2024)03—0025—11
sRNA 是植物发育和种子形成的重要调节因子。植物中的sRNA 包括short interfering RNA(siRNA) 和microRNA(miRNA), 其中,siRNA 包括ra-siRNA(重复相关小干扰RNA)、天然反义转录物衍生的小干扰RNA(naturalantisense transcript-derived small interfering RNA,natsiRNAs)、反式作用反义小分子干扰RNA(transactingantisense interfering RNAs,ta-siRNAs)、异染色质小分子干扰RNA(heterochromatic smallinterfering RNA,hc-siRNA)、初级siRNAs(PrimarysiRNAs) 和长的小分子干扰RNA(long smallinterfering RNAs,lsiRNA)[1-2]。其中,miRNA 是一类真核生物中广泛存在长度约为20 ~ 24 个核苷酸(nucleotide,nt)的内源非编码单链RNA,它通常是由RNA 聚合酶Ⅱ合成,其最初的转录产物称初始miRNA(pri-miRNA 初级转录产物),初始miRNA 被DCL(Dicer-like)酶切割成双链premiRNA(precursor miRNA),它通过碱基互补直接作用于靶基因mRNA,诱导靶基因的特异性切割,在转录和转录后水平调节基因的表达[3-4]。如,从马铃薯叶、花、根、块茎的sRNA 表达图谱发现sRNA 进化率低并且较为保守,具有组织特异表达的特性[5],研究还发现:在龙眼花发育过程中,花芽中的sRNA 富集表达,通过信号途径参与花器官发育转录调控网络的特异调控[6];在鹅掌楸叶和花中,miR157a-SPL 和miR160a-ARF 等miRNA差异表达[7]。因此,sRNA 在植物营养生长、生殖生长[8-9]、油脂形成[10] 等发育过程和模式中发挥重要作用[11]。
高通量测序由于可以同时测序几十万到几百万条RNA 分子,读取较短序列,检测全面、灵敏度高,因而加快了sRNA 的发现和功能研究[12]。目前,已经从179 种植物中鉴定出38 186 个miRNA 和7 838 个家族[13-15]。在拟南芥中miRNA参与胚形成并且抑制靶基因的表达,调控胚成熟[16]。如,在胚缺失突变体lacking DCL1 八细胞胚期, 上调较多的是miR156 靶基因、编码SPL10 和SPL11 的转录因子。在胚成熟过程中,miRNA156 诱导介导抑制zygotic SPL 转录, 抑制转录的近成熟积累[16]。Lin 等[17] 以龙眼的同步胚性培养物为材料,研究体细胞胚胎发生过程的miRNA 和靶基因,发现了272 个miRNA 和2063个靶基因,其中,181 个miRNA 和862 个靶基因得到证实。这些靶基因参与植物代谢、信号传导和应激响应。研究还发现:20 个保守miRNA 和4个新miRNA 参与体细胞胚状体的发育过程。在油菜Brassica napus 中发现了50 个保守miRNA,其中,一些miRNA 影响油脂的形成积累[10]。然而,对多数sRNA 的表达和功能仍然了解甚少。本研究以油茶叶片和种子为材料构建sRNA文库,利用Illumina高通量测序技术,确定已知miRNA 和新miRNA,筛选差异表达miRNA,预测靶基因,进行靶基因GO功能富集分析和KEGG通路富集分析,揭示miRNA 与光合作用、碳固定和油酯代谢过程中对应靶基因间可能的调控关系,为利用miRNA 技术进行油茶遗传改良提供科学依据。
1 材料与方法
1.1 RNA提取和构建sRNA文库
采集油茶‘横冲89’的嫩叶和成熟种子,立即液氮冷冻,-70 ℃贮藏。利用CTAB 裂解法提取总RNA。以总RNA 为起始样品,利用SmallRNA Sample Pre Kit,将Small RNA 加上接头反转录合成cDNA,PCR 扩增,分离回收DNA,构建cDNA文库。利用Qubit2.0 和Agilent 2100对文库的insert size 进行定量检测和完整性分析,采用HiSeq/MiSeq2000测序获得原始序列。
1.2 测序数据质量评估和sRNA的结构特征、种类和数量、公共序列及特有序列分析
通过Phred数值,转化获得碱基测序错误率(e),每个碱基测序错误率通过公式Qphred=-10log10(e)转化得到,以Phred 分值及对应的Q20、Q30评估数据质量。去除原始读长(raw reads)中低质量、含N、5′ 接头污染、无3′ 接头序列、polyA/T/G/C的读长,得到净读长(clean reads)。在18~40nt 的长度区间筛选sRNA,分析sRNA 的长度分布、种类、数量以及公共及特有序列等。
1.3 已知miRNA 分析、新miRNA预测以及miRNA家族分析
用bowtie 把sRNA 定位到拼接的油茶转录组序列上,将mapped 到参考序列上的sRNA 序列与miRBase 序列比对,比对分析已知miRNA,分析比对上的总sRNA 种类、数量等[18-19]。分析已知miRNA 首位碱基的偏好性。利用miREvo[15]和mirdeep2[20]miRNA 软件,通过miRNA 的发夹结构,分析比对上miRNA 前体和成熟体,预测新miRNA(novel miRNA),对miRNA 进行家族分析,探索miRNA 家族在其他物种中的存在情况。
1.4 miRNA表达、差异分析和聚类分析
从miRNA 表达水平分析中得到的read count数据,采用TPM对read count 数据进行归一化处理,归一化表达量= Mapped read count/Totalreads×1 000 000。利用DEGseq(2010)R package进行差异分析。用q value 调整P-value,以q value <0.01 及|log2(fold change)| > 1作为默认阈值进行显著差异性分析。将差异miRNA 的TPM 值用于聚类分析。
1.5 miRNA靶基因预测、候选靶基因GO 功能富集分析
和KEGG 通路富集分析通过psRobot_tarin psRobot 预测miRNA 靶基因。利用GO(Gene Ontology,http://www.geneontology.org/)数据库对差异表达miRNA 靶基因进行功能富集分析,将差异表达miRNA 的候选靶基因按照生物学过程、细胞组分、分子功能进行富集条目(term)分析。通过KEGG(KyotoEncyclopedia of Genes and Genomes)通路显著性富集,分析差异表达 miRNA 候选靶基因参与的主要代谢途径和信号转导途径。
1.6 参与光合作用、油酯代谢miRNAs 的表达分析和靶基因筛选
根据miRNA 的差异表达、候选靶基因的KEGG pathway 富集分析,筛选油茶光合作用、脂肪酸油脂代谢通路的miRNA 靶基因,预测差异表达miRNA 与靶基因可能的调控关系。
2 结果与分析
2.1 总RNA 提取与sRNA 文库制备
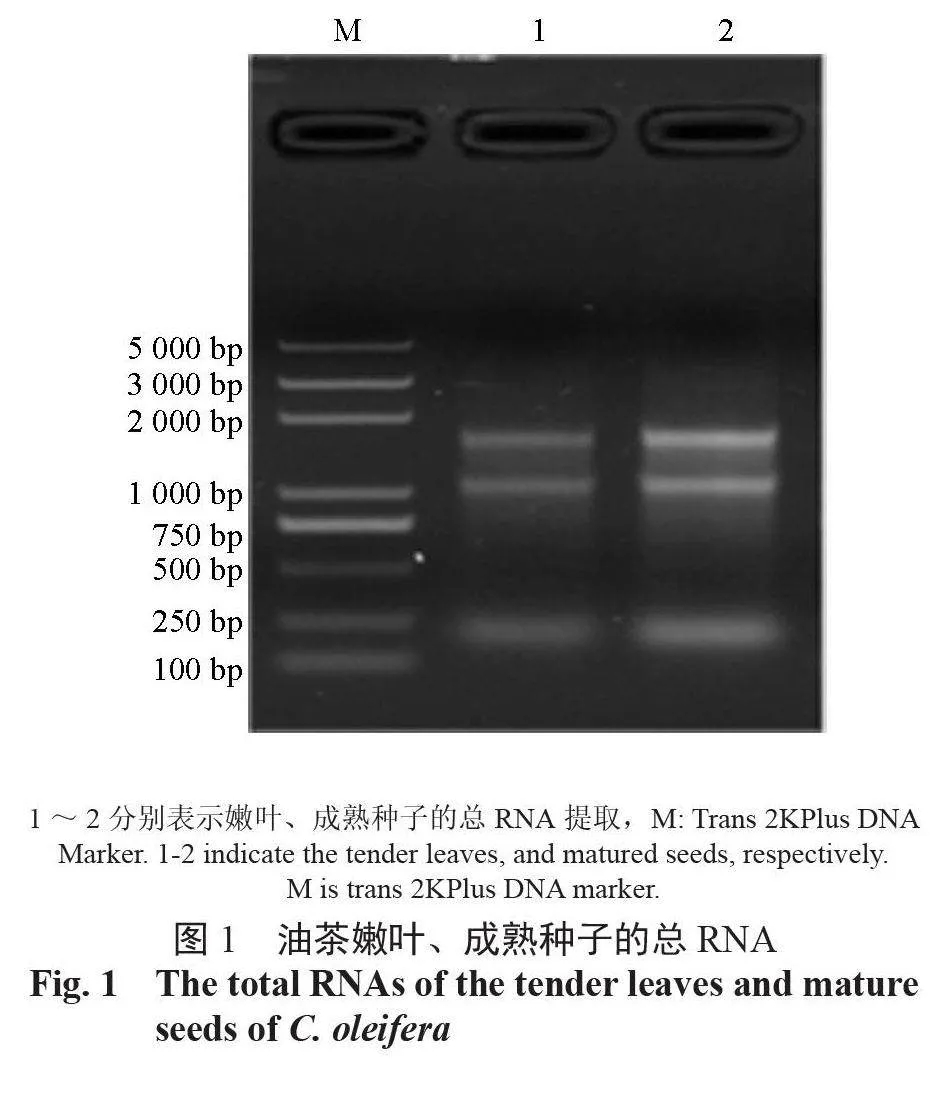
提取油茶幼叶和成熟种子的总RNA,用DNase Ⅰ除去残留的基因组DNA(图1)。RNA浓度A260/A280 分别为2.119 和2.093。总RNA 浓度达到了建库要求,Agilent 2100 Bioanalyzer(AgilentTechnologies,USA)测定RNA 完整性,结果表明:RIN(RNA integrity number) 为8.2 和8.9, 符合建库标准。分离富集叶片和种子的sRNA,构建sRNA 文库。
2.2 测序数据质量评估和sRNA 的结构特征、种类和数量
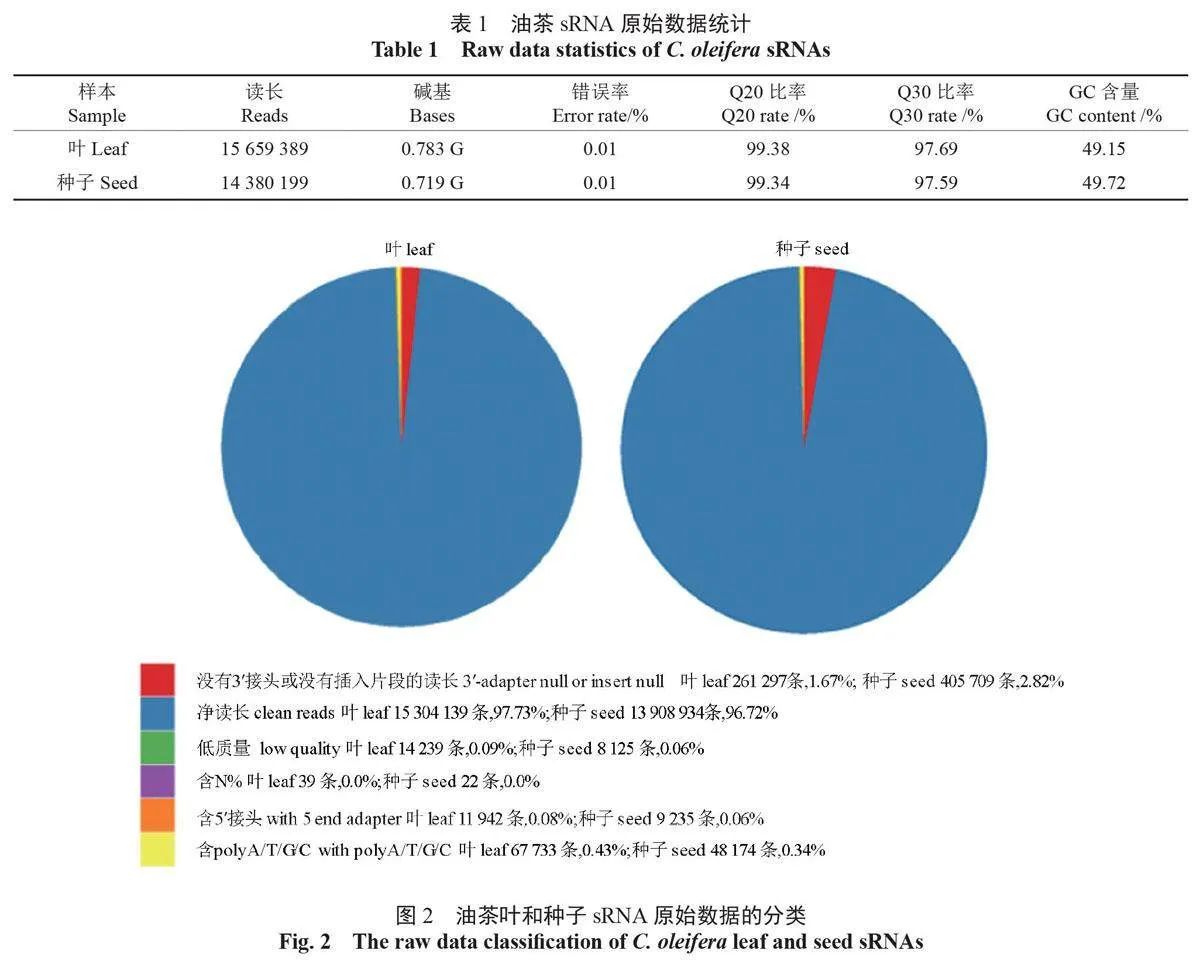
通过测序获得油茶叶sRNA 的原始读长为15 659 389 条,测序错误检验的错误率为0.01%,Q20、Q30 分别达到99.38% 和97.69%,碱基GC 含量为49.15%;种子sRNA 的原始读长为14 380 199条,测序检验错误率为0.01%,Q20 和Q30 分别达到99.34% 和97.59%,碱基GC 含量为49.72%(表1)。
在叶和种子sRNA 中,无5′ 接头或无插入片段读长分别为261 297 条(1.67%)和405 709 条(2.82%);低质量读长分别为14 239 条(0.09%)和8 125 条(00.6%)、含N% > 10% 的读长分别为39条和22条;含5′ 接头的读长分别为11942 条(0.08%)和9,253 条(0.06%),含有polyA/T/G/C的读长分别为67733条(0.43%)和48174条(0.34%),除去上述读长,获得叶和种子sRNA 的净读长分别为15304139条和13908934条(图2)。
在筛选获得油茶叶和种子的sRNA 中,18 ~ 40 nt 的净读长数量分别为14 256 189 条和10 822 313条,属于6 138179和4980518 种sRNA。与其他植物相似,油茶sRNA 的长度分布在18 ~ 40 nt,其中,24-nt sRNA 最多,在叶和种子中分别为4678323条(32.82%)和3 516 310条(32.49%);其次是21-nt sRNA,分别为954 749条(6.7%) 和1142946 条(10.56%);22-ntsRNA 居于第3,分别为952210条(6.68%)和941611(6.12%)条;23-nt 的居于第4,分别为872 524 条(8.7%)和749 356 条(6.92%)(图3)。油茶叶和种子sRNA 公共序列的种类为773647种(7.48%),特有序列的种类分别为4206871种(40.67%) 和5364532种(51.86%);公共序列的数量为1489104条(59.38%)。叶和种子sRNA 特有序列的数量分别为4418438条(23.0%)和5768160条(17.62%)。
2.3 已知miRNA 分析、新miRNA预测以及miRNA家族分析
用bowtie 将sRNA 定位到trinity 拼接的转录组上,结果发现:叶和种子中的sRNA 分别为14 256 189 条和10 822 313 条,比对到参照序列的sRNA 分别为5 855 009 条(41.07%)和4 122 070条(38.09%)。其中,与参考序列同向的分别为933 734(6.55%) 条和770 311 条(7.12%), 与参考序列反向的分别为4 921 275(34.52%)条和3 351 759 条(30.97%)。比对分析已知miRNA,结果发现: 比对上的总sRNA(Total sRNA)为120 102 条,在叶和种子中分别为54 035 条和66 067 条;比对上的sRNA 为518 种,在叶和种子中分别245 种和273 种;比对上的miRNA 前体为118种,其中,在叶和种子中分别为112 种和109 种;比对上的miRNA 成熟体为78 个,其中,在叶和种子中分别为74 个和72 个。其中,ath-miR399d、osa-miR399e、ath-miR399f、gma-miR169v、osamiR1863a、osa-miR1863b 的成熟体仅出现在叶中,而ath-miR171a、ath-miR5658、gma-miR172b-5p、osa- miR1863b 的成熟体仅出现在种子中。
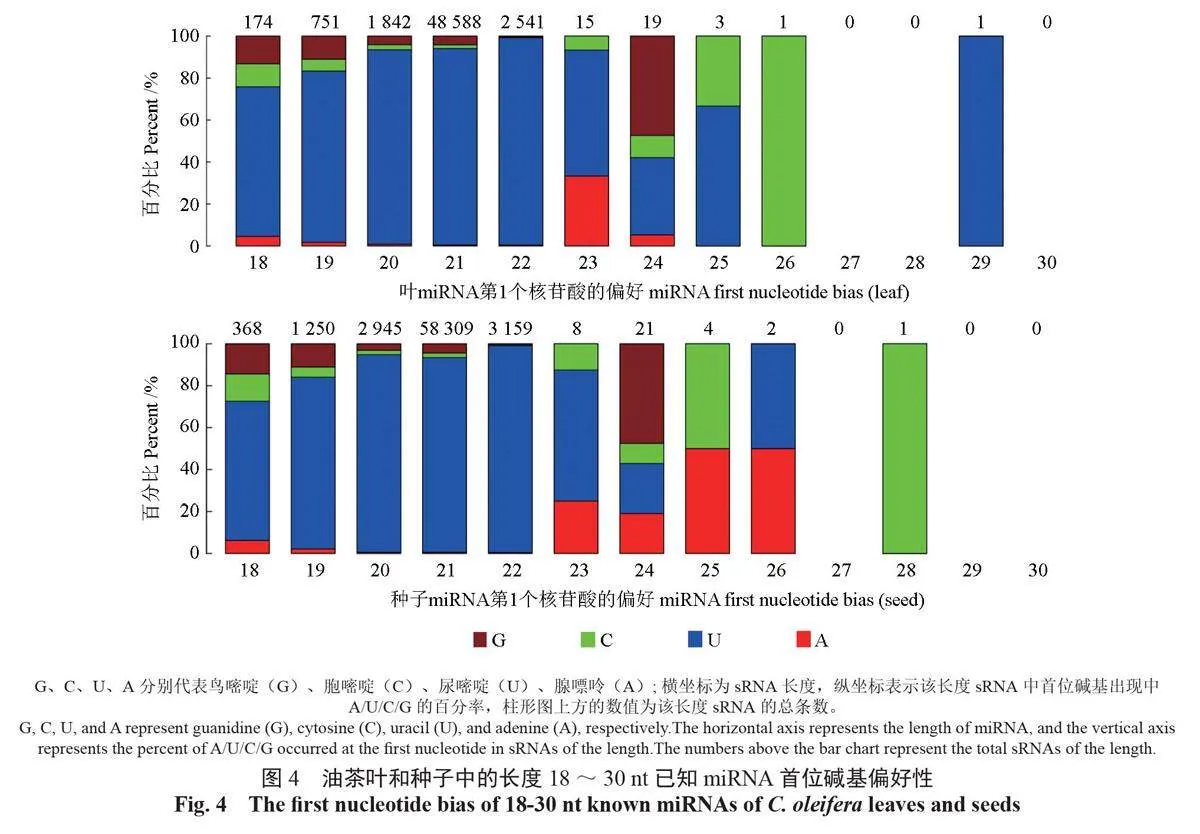
从图4可以发现:18-23nt 的已知miRNA 首位碱基多偏好碱基U(uridine), 其中,21-nt 已知miRNA 的首位碱基为U 的sRNA 数量最多,分别为45 579 条和54 137 条;22-nt 已知miRNA的次之,分别为2510条和3118条;20-nt 已知miRNA 的为第3位,分别为1707条和2775条。
利用miRDeep2软件进行新miRNA(novelmiRNA)预测,结果发现:比对上新miRNA 前体的sRNA 种类为1178种,总sRNA 为20653个。其中,在叶中比对上的sRNA 种类为596种,总sRNA 为10330个;在种子中为582种,比对上前体的总sRNA 为10323个。分析发现了54个新miRNA,长度为19-24nt,以24-nt miRNA 为主。在叶片中比对上的新miRNA 成熟体出现了9 411次,在种子中的出现了9 504 次。此外,发现33个新miRNA 含有miRNA*(miRNA 的随从链),在叶和种子中miRNA* 分别出现了443 次和464次(表2)。
2.4 miRNA表达、差异分析
和层次聚类分析统计分析miRNA 的读长数(read count),TPM 归一化处理这些读长数,筛选出132 个差异表达的miRNA,其中,在叶中为128 个,在种子为126 个,122 个为两者共表达,叶和种子表达的sRNA 读长129 568 条和154 492 条。其中,ath-miR166a 对应读长最多,在叶和种子中分别出现了26 594 次和29 821 次。DEGseq 差异分析发现:在叶和种子中有26 个显著差异表达的miRNA(q value < 0.01,|log2(fold change)| > 1),显著上调和下调表达的miRNAs 均为13 个。其中,显著上调表达的miRNA 包含ath-miR169b、ath-miR399d、ath-miR399f、gma-miR169v、osamiR1863a、osa-miR399e 6 个已知miRNA 和novel82、novel 32、novel 54、novel 49、novel 53、novel 41、novel 83 6 个新miRNA。其中,athmiR169b、novel 82、ath-miR399d、ath-miR399f、gma-miR169v、osa-miR1863a、osa-miR399e 的log2 倍数差异表达值大于3。显著下调表达的miRNAs 包括ath-miR172e、gma-miR172k、gmamiR396b-3p、gma-miR172c、gma- miR172d、ath-miR172c、ath-miR399b、osa-miR172c、gmamiR172b-5p、ath-miR171a、ath-miR5658、osamiR1863b12 个已知miRNA 和novel 72。其中,gma-miR172b-5p、ath-miR171a、ath-miR5658、osa-miR1863b 的log2 倍数差异表达值小于-3。
由图5 聚类分析可知:在叶和种子miRNA 表达模式较为相似。差异表达的miRNA 被分为2 族,一族是在叶和种子中的表达量均较低,包括athmiR5658、osa-miR1863b、ath-miR171a 等。另一族是在叶和种子中的表达量较高,包括ath-miR399b、ath-miR172c、gma-miR369b-p、novel 53、novel 32、novel 54 等。另外,novel 49、novel 41、novel 82 在仅叶中表达量高;osa-miR172c、gma-miR172c 和gma-miR172d 等在仅种子中表达量高。
2.5 miRNA靶基因预测、候选靶基因GO功能富集分析和KEGG通路富集分析
对差异表达的miRNA 进行靶基因预测,得到67个差异表达miRNA 和它们对应的1346个靶基因。26 个显著差异表达的miRNA 对应的靶基因为644个。结果还发现:一个miRNA 调控多个靶基因,如:ath-miR5658调控532个靶基因;同时,一个靶基因受多个miRNA 调控,如调控胚胎发育与细胞增殖的真核翻译起始因子3类-L- 亚基基因(comp69386_c0) 受到ath-miR156g、athmiR156j、osa-miR156k 3 个miRNA 的调控。
根据差异miRNA 候选靶基因在GO 数据库中的分布特征,计算显著富集的GO条目(term)。从图6可以看出,叶和种子差异表达miRNA 的候选靶基因在生物学过程、细胞组分、分子功能中的51 个条目中富集。其中,生物学过程中的生物调控过程、细胞过程、代谢过程等23 个条目富集较多;细胞组分中的细胞、细胞部分、细胞器、细胞器等17个条目富集较多;分子功能过程的结合、催化活性等11个条目显著富集。KEGG 通路富集分析结果表明:候选靶基因在萜类主链生物合成、甘油酯代谢、氨酰tRNA 生物合成、RIG-I样受体、核黄素代谢、角质软木脂和蜡质生物合成等条目中显著富集。
2.6 参与光合作用、油酯代谢miRNAs的表达分析和靶基因筛选
从表3可以发现:ath-miR5658 在叶片中没有表达,而在种子中表达量为6.47。它对应靶基因的靶基因分别是光合碳固定、光合作用途径中的类NADP 依赖型苹果酶(comp89613_c0)、果糖二磷酸醛缩酶1(comp46464_c0)、光系统I 反应中心亚基ⅣB(comp71063_c0),甘油磷酸酯代谢中的甘油-3- 磷酸酰基转移酶1(comp77968_c0),脂肪酸延长途径中的3- 酮酰基-CoA 合成酶4(comp80917_c1),甘油脂代谢和甘油磷酸酯代谢中的1- 酰基-sn- 甘油-3- 磷酸酰基转移酶5comp83553_c0 等。而gma-miR169v 在叶片中的表达量为7.72,而在种子中没有表达。它的靶基因是甘油脂代谢中的单半乳糖酰二酰基甘油合酶2(comp90506_c0)。
3 讨论
3.1 sRNA的数量、长度、种类鉴定和特征分析
sRNA 是植物发育和种子形成的重要调节因子。通过长度分布能够鉴定sRNA 的种类,如,21~24 nt 的sRNA 主要是miRNA,多数植物的siRNA(small interfering RNAs) 为24 nt [2,17,26]。研究发现:油茶叶和种子中sRNA 最多的是24-ntsRNA,其次是21-nt sRNA。这与油茶抗炭疽病[27]略有差别,与Brassica napus[10]、Arabidopsis thaliana、Oryza sativa[28] 的sRNA 相似,第3、4 大分布群体是22-nt 和23-nt sRNA,在叶种子中分别占6.68%和6.12%;在种子中占8.7% 和6.92%(图3)。
3.2 miRNA 的碱基偏好特征分析
植物miRNA 的5′ 端首个碱基偏好U 而抵抗G。可以通过miRNA 的U 碱基偏好性分析,获得典型性miRNA 序列比例。从图4 可以发现,长度在18-23 nt 的已知miRNA 首位碱基偏好U,这与中国甜柿[29] 中的miRNA 有一定的相似性,因此,增强了研究鉴定结果的可信度。
3.3 已知miRNAs家族分析、新miRNA 鉴定和靶基因分析
本研究从油茶叶和种子sRNA 中鉴定出22 个已知miRNAs 家族,105 个miRNAs 家族成员。其中,多数已知miRNA 与其他物种保守miRNA 具有高度相似性[27]。在鉴定出的miRNA 家族中,以多家族成员形式存在多个物种,这与相关研究结果[27] 相一致。本研究初步发现和证实了“一个miRNA 可能存在多个靶基因,而多个miRNA 可能均调控同一靶基因”的现象[27]。其中,67 个表达差异miRNA 调控1 346 个靶基因。在油茶中发现54 个油茶新miRNA,其长度以24 nt 为主,新miRNA 的数量和长度与杨利利[27] 对油茶的研究结果相一致,但与中国甜柿新miRNA 的长度分布(以21 nt 为主)略有差异[29]。筛选出的miRNAs 及其候选靶基因还有待进一步利用实时荧光定量PCR和特殊茎环qRT-PCR 进行验证。由于缺少油茶的基因组序列,本研究无法进行茎环结构准确预测,导致所得的miRNA 可能是其他类型的sRNA。此外,由于缺少基因组注释,可能会导致靶基因注释不全。但是,随着油茶的核基因组、叶绿体基因组、线粒体基因组等基因组组装和测序解析研究的深入[30],sRNA 种类的预测,特别miRNA 会更加准确。
3.4 调控光合作用和油酯代谢miRNA靶基因的表达分析
从表3 可以发现:ath-miR5658 在叶片中没有表达,而在种子大量表达。而gma-miR169v 在叶片中大量表达,而在种子中没有表达。前期的转录表达研究结果[19] 显示:类NADP依赖型苹果酶(comp89613_c0)、果糖二磷酸醛缩酶1(comp46464_c0)、光系统I反应中心亚基ⅣB(comp71063_c0)、单半乳糖基二酰基甘油合酶2(comp90506_c0)等基因在油茶叶中的转录表达较种子中均上调表达。这表明:ath-miR5658 没有显著抑制这些靶基因的转录表达,对油茶叶片光合产物碳固定、光合作用的影响较小。而靶基因甘油磷脂代谢1- 酰基-sn- 甘油-3- 磷酸酰基转移酶5(comp83553_c0)、甘油-3- 磷酸酰基转移酶1(comp77968_c0)、3- 酮酰基-CoA 合成酶4(comp80917_c1)在叶和种子中的转录表达没有明显差异,ath-miR5658 可能调控了这些靶基因在种子中的转录表达。据此我们推测:ath-miR5658既可能调控叶片中光合作用基因的表达[20],但也会对种子中的甘油磷酸酯代谢、脂肪酸延长和甘油酯代谢产生影响。而gma-miR169v 尽管在叶片中表达,但是,其靶基因半乳糖基二酰基甘油合酶2(comp90506_c0)在叶片中的转录表达仍然远高于种子中的表达。由此可见,它的调控作用局限在一定范围内。
在前期油茶转录组表达分析研究[19] 的基础上,本研究进行miRNA 靶基因筛选, 初步确定了mRNA 和靶基因的调控关系。下一步研究将是利用基因组测序组装序列,stem loop qRT-PCR 和靶转录表达方法,一是验证ath-miR5658 及其靶基因类NADP 依赖型苹果酶(comp89613_c0)、果糖二磷酸醛缩酶1(comp46464_c0)的转录表达的相关性,从而确认miRNA 对靶基因的调控程度。二是在油茶果实发育和茶油形成不同阶段,检测gma-miR169v 及其靶基因的表达模式,串联分析揭示miRNA-mRNA 调控关系。探讨油脂合成是否直接或间接地受到多种miRNA 家族的调控。
4 结论
本研究构建了油茶叶和种子的sRNA 文库,鉴定出54个新miRNA,22 个已知miRNA 家族,包括105 个miRNA 家族成员;67个差异表达miRNA, 调控1346个靶基因, 其中,26个miRNAs 为显著差异表达,调控644 个靶基因。初步证实了“一个mi RNA 调控多个靶基因,以及多个miRNA 调控一个靶基因的现象”;发现了athmiR5658可能调控碳固定代谢、光合作用、甘油磷酯代谢、甘油酯代谢、脂肪酸延长等途径中靶基因的表达,gma-miR169v 可能调控甘油酯代谢中靶基因的表达。
