
中国人群33 104 例单基因病携带者筛查的多中心研究
2024-11-03 00:00:00侯伟付晓琳谢潇潇张春燕边佳昕毛翛文娟罗春玉金华祝茜戚庆炜钱叶青袁静赵彦艳尹爱兰李树铁蒋宇林张蔓丽肖锐卢彦平
南方医科大学学报 2024年6期
摘要:目的 通过大规模多中心的多种遗传病携带者筛查,调查中国人群单基因病的流行病学特征以及突变谱,为制定适合中国人群的遗传病预防策略提供依据。方法 本研究在中国的12个临床中心共招募33 104例受检者(16 610例女性),基于高通量测序和多种PCR对223个基因的携带者状态进行检测。结果 197个常染色体基因的合并携带者频率为55.58%,26个X连锁基因的合并携带者频率为1.84%。在16 669 例家系中,共检出874 对(5.24%)高危夫妇。其中常染色体基因高危夫妇584 对(3.50%),X连锁基因高危夫妇306对(1.84%),16对夫妇同时为常染色体基因和X连锁基因高危夫妇。最常检出的常染色体高危基因包括GJB2(常染色体隐性耳聋1A,393 对),HBA1/HBA2(α-地中海贫血,36 对)和PAH(苯丙酮尿症,14 对),SMN1(脊髓性肌萎缩症,14对)。最常检出的X连锁高危基因包括G6PD(G6PD缺乏症,236对),DMD(进行性假肥大性肌营养不良,23对)和FMR1(脆性X综合征,17对)。除外G6PD后的高危夫妇率为3.91%(651/16 669),进一步除外GJB2 c.109Ggt;A位点后,高危夫妇率为1.72%(287/16 669)。理论上严重的单基因病出生缺陷的发病率约为4.35‰(72.5/16 669)。对导致高危夫妇最多的22 个基因进行筛查可检出95%以上的高危夫妇,对导致高危夫妇最多的54 个基因进行筛查可检出99%以上的高危夫妇。结论 本研究揭示了我国人群中223种单基因病的携带者频率,为中国人群的携带者筛查策略制定和panel设计提供依据。在携带者筛查实践中,针对某些特殊基因或变异位点的遗传咨询可能会面临困难。这些特殊基因或变异需要在检测前告知受检夫妇,并在可能的情况下提供不筛查这些基因或变异的选择。
关键词:携带者筛查;单基因病;遗传咨询
单基因病是先天性缺陷的主要原因之一,也是影响公共卫生的全球性问题[1]。无症状的单基因病携带者有可能生育患儿。既往研究已发现7000 余种单基因病[2],其中有严重临床症状的常染色体隐性遗传病和X连锁遗传病约1300余种[3],有研究显示严重隐性遗传病的累计发病率为1/500[4]。携带者筛查的目的是在普通人群中识别携带致病基因的无症状个体,评估其后代患遗传病的风险,通过进一步的遗传咨询和生育指导,避免严重遗传病的发生[5]。
单基因病的携带者频率是一项重要的流行病学指标,为临床遗传咨询和公共卫生决策提供重要依据。最早的遗传病携带者筛查是在德裔犹太人中对Tay-Sachs病进行的筛查,这一实践正是因为在该人群中发现了异常高的携带者频率[6]。随着科技的进步,特别是高通量测序技术的普及和测序成本的降低,携带者筛查的目标人群和疾病范围也相应扩展。美国妇产科医师学会(ACOG)和美国医学遗传学和基因组学学会(ACMG)均将携带者频率作为选择筛查疾病的标准之一,这进一步强调了其在遗传病预防和管理中的重要性[7, 8]。准确地了解和利用携带者频率数据,对于计算筛查的残余风险、提供有效的遗传咨询以及制定合理的筛查策略都具有至关重要的意义[9, 10] 。
携带者筛查已进入多基因同时筛查的扩展性携带者筛查时代。然而,尽管单基因病的携带者筛查在全球范围内已经得到了广泛的关注和实践,但目前仍缺乏针对中国人群的大规模、多中心的携带者筛查研究。以往的中国人群携带者筛查研究往往局限于较少的病种数量,如遗传性耳聋和地中海贫血,或者仅在中国的局部地区进行,难以全面反映中国人群中单基因病的流行特征[11-14]。公开的人群频率数据库中也缺乏对中国人群的代表性[15]。加之我国人口众多,经济发展不平衡,遗传咨询能力不足,如何开展适合国情的携带者筛查是一个迫切需要解决的问题。
基于以上背景,在国家重点研发计划支持下我们开展了这项包含33 104例中国人群的单基因病携带者筛查研究。期望通过这一大规模、多中心的临床研究实践,揭示中国人群中单基因病的流行病学特征以及突变谱;结合实施过程中发现的问题,为制定适合中国人群的遗传病预防策略提供科学依据。
1 资料和方法
1.1 受试者招募
2022年7月20日~2023年10月31日,本研究在中国的12 个临床中心共招募33 104 例受检者(16 610 例女性)。入组标准:备孕或孕周小于等于13+6周的单胎妊娠的夫妻;签署书面知情同意书。排除标准:辅助生殖受孕;已经被诊断为单基因疾病患者或携带者;在1年内接受过输血、器官移植或免疫治疗。本研究已通过伦理审查委员会审核批准[中国人民解放军总医院医学伦理委员会(伦审第S2021-474-01 号),中南大学医学伦理委员会(伦审第202107009号),济南市妇幼保健院伦理审查委员会(伦研批第2021-1-036),南京市妇幼保健院医学伦理委员会(宁妇伦字2021 NFKSL-086号),中国医科大学附属盛京医院医学伦理委员会(2021PS661K),四川大学华西第二医院医学伦理委员会(2021 伦审批第155 号);湖南省妇幼保健院伦理委员会(快202160 号),浙江大学医学院附属妇产科医院医学伦理委员会(IRB-20210242-R),安徽医科大学第一附属医院临床医学研究伦理委员会(伦审-快-PJ2020-06-51)等]。
1.2 筛查疾病的选择
本研究共对223个基因进行筛查。筛查的疾病根据如下标准选择:(1)已知携带频率为1/100或更高;(2)具有明确的表型;(3)对生活质量有严重不良影响;(4)导致认知或身体损害;(5)需要医疗或手术干预;(6)生命早期发病;(7)改变分娩管理以改善新生儿结果,并教育父母了解出生后的特殊护理需求。由于部分基因在中国人群中的携带者频率尚不明确,本研究的疾病列表基于4个来源:(1)文献研究:一般人群中的高频变异以及中国或东亚/东南亚人群中的常见变异;(2)Gnomad 数据库:东亚/东南亚中的高频LOF变异;(3)其他专业数据库,如HGMD、ClinVar和LOVD;(4)本地数据库。
1.3 文库制备及高通量测序
本研究使用通用下一代测序文库制备试剂盒(Biosan Tech)进行DNA文库制备。使用文库定量试剂盒(Roche)按照生产商说明对纯化的DNA文库进行定量。本研究使用定制的携带者筛查捕获探针试剂盒(Biosan Tech)。该探针区域覆盖了目标基因所有外显子和已报道的内含子中的致病性(P)/可能致病性(LP)变异。目标区域包括所有单核苷酸变异(SNV)、10 bp以内的插入/缺失变异,以及部分缺失/重复变异。利用捕获探针试剂盒,结合目标区域捕获和高通量测序方法,在MGI-DNB-T7 测序仪上对样本进行测序。将获得的DNA 序列与NCBI 人类参考基因组(hg19/GRCh37)进行比较。检测到的变异进行了生物信息学分析和致病性解读,最终报告P和LP变异。此外,X连锁基因的检测限于女性受检者。
1.4 FMR1基因检测
使用脆性X综合征CGG重复数检测试剂盒(PCR -毛细管电泳法)(Biosan Tech)检测FMR1相关的脆性X综合征。该试剂盒通过基因特异性PCR和三重重复引物PCR偶联扩增,计算外周血样本中FMR1基因5'端非编码区CGG重复序列的数量和范围。这项测试仅限于女性受试者。
1.5 F8基因倒位检测
采用血友病F8 基因检测试剂盒(PCR -电泳法)(Biosan Tech)。该试剂盒采用LR-PCR结合琼脂糖凝胶电泳检测F8基因内含子1和内含子22的倒位。这项测试仅限于女性受试者。
1.6 携带者频率的计算
计算携带者频率时仅考虑P/LP变异的携带者,不计入临床意义不明变异(VUS)的携带者。单一受检者在同一个基因上检出多个P/LP变异时只计数1次,不会记作多个携带者。X连锁基因的携带者频率仅统计女性受检者。
1.7 检测前的遗传咨询
宣教扩展性携带者筛查的检测方法、目的、意义以及筛查和诊断流程,并介绍目前的研究项目以及入组后的获益及风险。
1.8 检测后的遗传咨询
安排专人发放科研检测报告并进行遗传咨询。疑难病例提交专家组讨论,必要时提交项目组建立的多学科会诊平台讨论。
2 结果
2.1 总体人群中的携带者情况
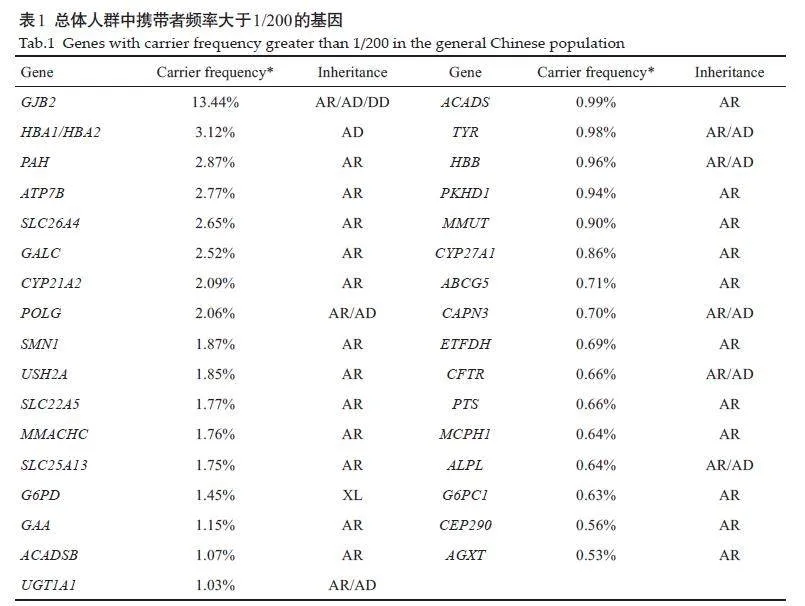
本研究。总体人群中,进行筛查的197 个常染色体基因的合并携带者频率为55.58%(18 399/33 104),26 个X连锁基因的合并携带者频率为1.84%(306/16610)。在18 527例携带者中,12 111例(65.37%)携带1个变异,4870 例(26.29%)携带2 个变异,1274 例(6.73%)携带3 个变异,245 例(1.32%)携带4 个变异,54 例(0.28%)携带5个及以上变异。总体人群中携带者频率最高的基因及对应疾病是GJB2(常染色体隐性耳聋1A,13.31%),显著高于其它基因;其它携带者频率较高的基因及对应疾病依次为HBA1/HBA2(α-地中海贫血,3.09%),PAH(苯丙酮尿症,2.79%),ATP7B(肝豆状核变性,2.75%)和SLC26A4(常染色体隐性耳聋4型,2.62%)。总体人群中共有17个基因的携带者频率大于1/100 34个基因的携带者频率大于1/200(表1)。
2.2 高危夫妇检出情况
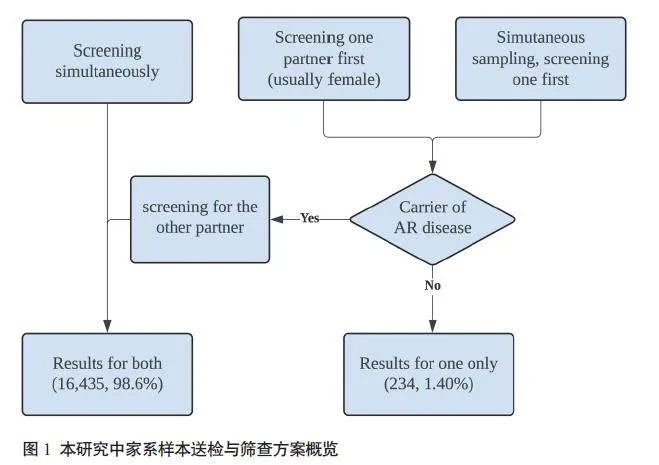
高危夫妇定义为夫妻双方为同一常染色体基因的携带者,或女方为X连锁基因的携带者。图1显示了本研究中的家系样本送检情况及筛查方案。本研究包含16 669例家系,其中16 435例(98.60%)家系送检夫妻双方样本,234 例(1.40%)家系仅送检一方样本。所有家系中共检出874对(5.24%)高危夫妇。常染色体基因高危夫妇584 对(3.50%),X 连锁基因高危夫妇306 对(1.84%),16对夫妇同时为常染色体基因和X连锁基因高危夫妇。除外G6PD后的高危夫妇检出率为3.91%(651/16 669)。同时,考虑到GJB2 c.109Ggt;A位点相关疾病的表型差异较大,并且其等位基因频率高达5.24%,进一步除外GJB2 c.109Ggt;A位点后,高危夫妇检出率为1.72%(287/16 669)。按照单个高风险基因有1/4的概率生育患儿进行估计,两个基因独立遗传时生育患儿的概率为43.75%[(1-3/4×3/4)×100%],理论上本研究可避免约72.5例遗传病患儿的出生,严重的单基因病出生缺陷的发病率约为4.35‰(72.5/16 669)。
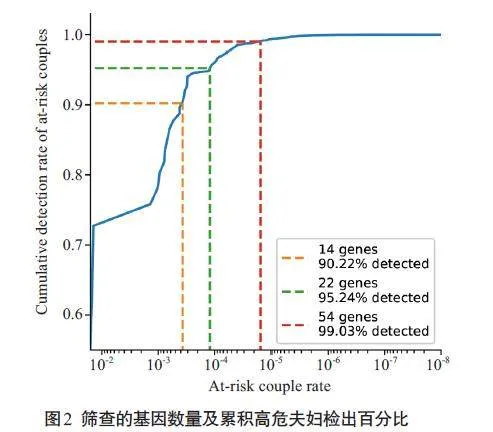
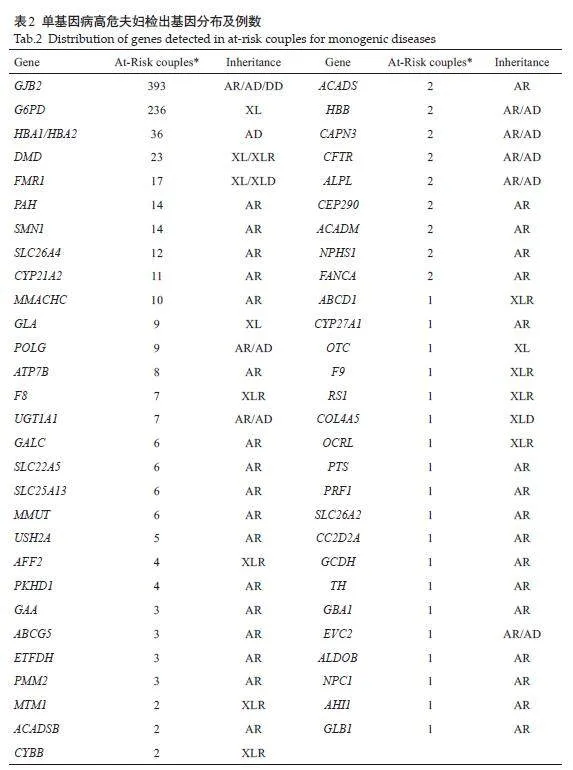
本研究共检出874对高风险夫妇,表2显示了检出的高危基因的分布及例数,共检出高危基因898次。在43 个常染色体基因和13 个X连锁基因上检出高危夫妇。最常检出的常染色体基因包括GJB2(常染色体隐性耳聋1A,393对),HBA1/HBA2(α-地中海贫血,36对)和PAH(苯丙酮尿症,14对),SMN1(脊髓性肌萎缩症,14对)。最常检出的X连锁基因包括G6PD(G6PD缺乏症,236对),DMD(进行性假肥大性肌营养不良,23对)和FMR1(脆性X综合征,17对)。图2显示只要对高危夫妇检出率较高的部分基因进行筛查,即可检出大部分的高危夫妇。对223个基因中高危率最高的54个基因进行筛查,即可检出99.02%的高危夫妇。
2.3 检出的致病性和可能致病性变异
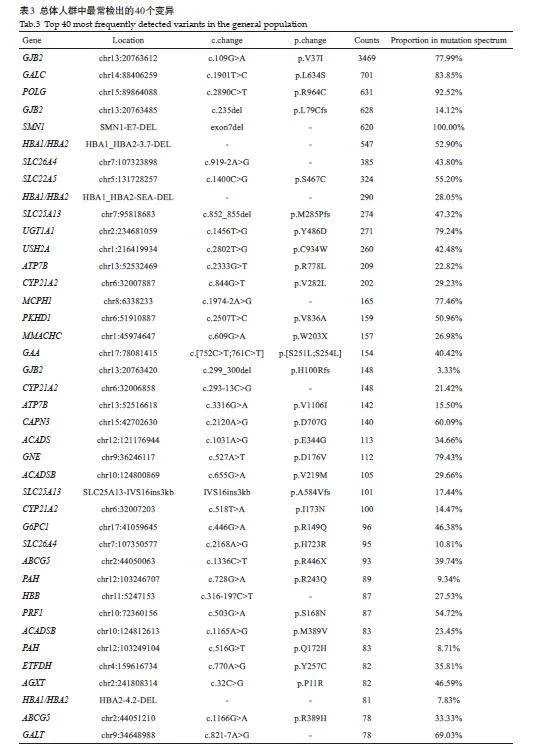
本研究在33 104例受检者的223个基因进行筛查,共检出5292种致病性或可能致病性变异,其中3405种(64.34%)变异仅检出过1次。表3显示了最常检出的40个变异位点。检出次数最多的位点为GJB2 c.109Ggt;A,共检出3469次,远高于其它位点。其它检出次数较多位点包括:GALC c.1901Tgt;C(701次),POLG c.2890Cgt;T(631次),GJB2 c.235del(628次)。
3 讨论
本研究是截止目前最大规模的中国人群携带者筛查研究,包含12个临床中心共计33 104例样本。本研究中197个常染色体基因的合并携带者频率为55.58%,26个X连锁基因的合并携带者频率为1.84%。2023年Chen等[13]对300对中国汉族夫妇进行携带者筛查,342种疾病的携带者频率为64.83%。2022 年Chau 等[11]对1543例中国南方人群进行315个基因的携带者筛查,携带者频率为47.8%,2022年Hu等[16]对1915对中国东部汉族夫妇进行24个基因中的448个致病变异位点筛查,携带者频率为18.5%。携带率的不同可能跟筛查的基因数量、采用的检测技术、筛查人群及变异报告原则等有关。本研究在16 669 例家系中,共检出874 对(5.24%)高危夫妇。除外GJB2 c.109Ggt;A及G6PD后,有287对(1.72%)高危夫妇需要孕前或产前干预以避免严重出生缺陷的发生。按照单个高风险基因有1/4的概率生育患儿进行估计,理论上本研究可避免约72.5例遗传病患儿的出生。严重的单基因病出生缺陷的发病率约为4.35‰(72.5/16 669),远高于唐氏综合征,由此可以看出在我国开展多个基因的携带者筛查对降低遗传病的首次发生具有重要意义。
本次筛查研究揭示了中国人群中一些常见的单基因病和变异位点,如GJB2基因相关的常染色体隐性耳聋、HBA1/HBA2基因关联的α-地中海贫血,以及GALC基因的克拉伯病等。这些相对常见的遗传病不仅对患者及其家庭造成深远影响,也给遗传咨询和产前诊断带来了挑战。因此,对于这些遗传病的深入学习和专业培训显得尤为重要。未来的遗传病携带者筛查咨询工作中,应加强对这些常见基因和变异的认知,提升医务人员的遗传咨询能力,以确保为受检者提供准确、全面的遗传信息和个性化的指导建议。
遗传病携带者筛查应包括多少基因一直是研究和争论的焦点。2017年,美国妇产科医师学会(ACOG)提出了携带者筛查基因选择的7项标准,其中携带者频率标准为大于1/100[8]。2021年,美国医学遗传学和基因组学学会(ACMG)指南提出的携带者频率标准为常染色体疾病大于1/200,X连锁疾病大于1/40 000[7, 10]。然而,虽然将更罕见的疾病纳入筛查的收益逐渐递减,但是并没有充足的证据来设定一条具体的携带者频率边界。增加筛查基因的收益相对明确,但是随之增加的成本却难以量化而且可变,主要包括变异判读的人工成本、遗传咨询的负担、产生的意外信息、个体作为特定疾病携带者的表型不确定性、焦虑等[17]。考虑到中国各地区间人工成本、遗传咨询能力、经济基础等差异以及不同的个体需求,有必要设计多种的panel以应对不同的情景[18]。本研究为中国人群的携带者筛查panel的设计提供了依据,基础的携带者筛查panel可考虑本研究中检出95%以上高危夫妇的22个基因,更高要求的筛查panel可以考虑99%(54个基因)的累积高危夫妇检出比。
一些不确定性因素,如疾病的低外显率和严重程度可变的表型等,会给携带者筛查的遗传咨询带来一定的困难。尽管ACMG和ACOG指南主要推荐针对那些具有严重临床表现且在生命早期就发病的疾病进行筛查,然而,由于等位基因异质性的存在,某些疾病可能展现出从轻微到严重的连续表型谱,或可能在个体的不同生命周期阶段出现发病情况。在本研究队列中,两个常见的变异是GJB2 c.109Ggt;A非截断变异(NT)和GJB2 c.235delC截断变异(T),携带率分别为10.48%和1.90%。这两个变异被ClinGen变异解释专家组归类为致病性[19]。基于截断和非截断变异的不同等位基因组合,可以观察到不同程度的听力损失。NT/NT基因型中53%的个体会出现轻度听力损失,13%的人有严重的听力损失;T/NT基因型中29%~37%的个体会出现轻度听力损失,24%~30%的人会出现严重的听力损失;T/T基因型有0~3%出现轻度表型,59%~64%出现严重听力损失[20]。这样的结果会给临床咨询带来巨大困难,造成不当引产,本研究经讨论后不在孕期夫妇中报告GJB2 c.109Ggt;A位点。此种处理方式必须在检测前后咨询时充分告知受检夫妇,以免后代出现严重表型带来医疗风险。本队列中GALC基因的c.1901Tgt;C变异位点的检出频率位列第二,其携带率为2.12%。半乳糖神经酰胺酶活性缺乏可引起克拉伯病。大多数的克拉伯病患者在1 岁以前发病,首先表现为极度易激惹、痉挛和发育迟缓。婴儿发病的克拉伯病的特征是最初几个月发育正常,随后迅速出现严重的神经功能恶化,平均死亡年龄为24月(8月~9岁)[21]。晚发性克拉伯病的临床表现和病程变化更大。GALC c.1901Tgt;C位点与晚发型克拉伯病相关[22-25]。c.1901Tgt;G突变患者的发病年龄从8个月到51岁不等[26]。婴儿期发病的变异几乎没有酶活性,而晚发型的酶活性为正常的4%~20%[24],但是酶活性与表型之间并没有明确对应的关系。除GJB2和GALC外,在其它基因上可能还存在类似的变异位点需要挖掘并予以关注。上述的特殊变异位点使得携带者筛查的遗传咨询和临床决策更加困难。理想情况下,这些表型预测困难的变异位点应当在检测前的遗传咨询中充分告知夫妇,并提供不筛查这些位点的选项。然而在大规模人口筛查及有限的遗传咨询能力的情况下,检测项目很难根据个人意愿进行调整,因此携带者筛查后的咨询需要针对具体的基因及变异针对性的咨询,以免引起不当的终止妊娠。
本研究将G6PD列入筛查,其在本队列的携带率高达1.42%。虽然G6PD缺乏症的患者大多数无症状,并且可能终生都不知道自己的疾病状态[27],但G6PD缺乏症可在新生儿中引起严重的间接高胆红素血症,可能导致核黄疸[28]。在受影响的新生儿中尽早诊断G6PD缺乏症可能有助于降低严重高胆红素血症、核黄疸的风险和减少输血的需要[29]。目前携带着筛查采用两种筛查方法,一种是基于测序的筛查,即本研究所采用的方法,另一种方法是基于基因分型的筛查,即仅筛查已知致病的变异。两种方法各有利弊,前者可检测目标范围内的所有变异,其中仅小部分是致病性和疑似致病性变异。这种方法可以发现更多罕见致病变异的携带者,但是检出的临床意义不明的变异也会给致病性判读和临床咨询带来困难。许多实验室仅报告P/LP变异,但是在夫妻一方为携带者的情况下,也可以考虑报告另一方同一基因内的VUS变异[7]。后者是定向筛查,筛查的结果是确定的。例如Hu等[16]基于毛细管电泳的多重PCR技术在中国东部人群中对24个基因中的448个致病变异进行检测,与测序为基础的携带者筛查相比,该方法成本低廉且结果易于解释,更适合遗传咨询能力较弱的机构或地区,然而该方法无法检出不常见的致病变异的携带者。此次大规模多中心的多基因筛查研究,为选择中国人群的热点突变提供了依据,未来可针对不同的受众设计不同的层级的筛查方案。
本项研究也存在一定的局限性。首先,尽管本研究对223个基因进行了筛查,但仍有可能遗漏了一些未引起重视但具有较高携带频率的基因。其次,本研究在招募受检者时未限定地区和民族,故未区分不同地区或民族间的携带频率差异,这通常会导致按照哈迪-温伯格平衡估计的高危夫妇率比真实观察到的偏低。为了更准确地评估遗传病风险,未来的研究应更加关注地区差异,并在筛查策略中充分考虑这些差异。最后,对于一些特殊基因的检测存在困难。这主要是由于特殊的变异类型和检测技术的局限性所致,例如CPY21A2、SMN1 等。大规模的基因转换可能会导致功能性的CYP21A2 序列被其同源假基因CYP21A1P片段替换,而假基因CYP21A1P 上存在多个有害变异[30]。SMN1exon7 缺失占其所有致病变异的95%~98%,序列突变占2%~5%[31, 32],加之SMN1的序列分析无法确定变异来源于SMN1 还是SMN2,因此对SMN1 的筛查仅限于对exon7缺失的检测。SMN1 exon7缺失的筛查可能存在假阴性,因为大约5%~8% 的人群染色体上有两个SMN1拷贝在一条染色体上,在另一条染色体上存在缺失,称为[2+0]基因型[33]。错误的致病性判读也可能导致一些真正的致病变异被遗漏。阴性结果并不能保证受检者不会生育患儿,携带者筛查呈阴性仍存在残余风险[34]。
综上所述,本研究不仅揭示了我国人群中单基因病的流行病学特征,更重要的是为制定适合中国人群的单基因病预防策略提供了依据。未来,随着技术的不断进步和数据的不断积累,有望进一步优化和完善筛查策略,大大降低我国严重遗传病的首次发生。
参考文献:
[1] Loh PR, Genovese G, McCarroll SA. Monogenic and polygenicinheritance become instruments for clonal selection[J]. Nature,2020, 584(7819): 136-41.
[2] Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severechildhood recessive diseases by next-generation sequencing[J]. SciTransl Med, 2011, 3(65): 65ra4.
[3] Henneman L, Borry P, Chokoshvili D, et al. Responsibleimplementation of expanded carrier screening[J]. Eur J Hum Genet,2017, 25(11): 1291.
[4] Haque IS, Lazarin GA, Kang HP, et al. Modeled fetal risk of geneticdiseases identified by expanded carrier screening[J]. JAMA, 2016,316(7): 734-42.
[5] Antonarakis SE. Carrier screening for recessive disorders[J]. NatRev Genet, 2019, 20(9): 549-61.
[6] Rose NC, Wick M. Carrier screening for single gene disorders[J].Semin Fetal Neonatal Med, 2018, 23(2): 78-84.
[7] Gregg AR, Aarabi M, Klugman S, et al. Screening for autosomalrecessive and X-linked conditions during pregnancy andpreconception: a practice resource of the American College ofMedical Genetics and Genomics (ACMG)[J]. Genet Med, 2021, 23(10): 1793-806.
[8] Committee opinion No. 690: carrier screening in the age of genomicmedicine[J]. Obstet Gynecol, 2017, 129(3): e35-40.
[9] Nussbaum RL, Slotnick RN, Risch NJ. Challenges in providingresidual risks in carrier testing[J]. Prenat Diagn, 2021, 41(9):1049-56.
[10] Johansen Taber K, Ben-Shachar R, Torres R, et al. A guidelinesconsistentcarrier screening panel that supports equity across diversepopulations[J]. Genet Med, 2022, 24(1): 201-13.
[11] Chau JFT, Yu MHC, Chui MMC, et al. Comprehensive analysis ofrecessive carrier status using exome and genome sequencing data in1543 Southern Chinese[J]. NPJ Genom Med, 2022, 7(1): 23.
[12]Fang YQ, Li JR, Zhang MM, et al. Clinical application value ofexpanded carrier screening in the population of childbearing age[J].Eur J Med Res, 2023, 28(1): 151.
[13]Chen SC, Zhou XY, Li SY, et al. Carrier burden of over 300 diseasesin Han Chinese identified by expanded carrier testing of 300 couplesusing assisted reproductive technology[J]. J Assist Reprod Genet,2023, 40(9): 2157-73.
[14]Zhao SM, Xiang JL, Fan CN, et al. Pilot study of expanded carrierscreening for 11 recessive diseases in China: results from 10, 476ethnically diverse couples[J]. Eur J Hum Genet, 2019, 27(2):254-62.
[15]Karczewski KJ, Francioli LC, Tiao G, et al. The mutationalconstraint spectrum quantified from variation in 141, 456 humans[J]. Nature, 2020, 581(7809): 434-43.
[16]Hu P, Tan JX, Yu F, et al. A capillary electrophoresis-based multiplexPCR assay for expanded carrier screening in the eastern HanChinese population[J]. NPJ Genom Med, 2022, 7(1): 6.
[17]Sparks TN. Expanded carrier screening: counseling andconsiderations[J]. Hum Genet, 2020, 139(9): 1131-9.
[18]中国妇幼保健协会生育保健分会, 黄荷凤, 徐晨明, 等. 针对生育人群的携带者筛查实验室和临床实践专家共识[J]. 中华生殖与避孕杂志, 2024, 10(2): 109-15.
[19]Shen J, Oza AM, del Castillo I, et al. Consensus interpretation of thep. Met34Thr and p. Val37Ile variants in GJB2 by the ClinGenHearing Loss Expert Panel[J]. Genet Med, 2019, 21: 2442-52.
[20]Huang SS, Huang BQ, Wang GJ, et al. The relationship between thep. V37I mutation in GJB2 and hearing phenotypes in Chineseindividuals[J]. PLoS One, 2015, 10(6): e0129662.
[21]Barczykowski AL, Foss AH, Duffner PK, et al. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomalstorage diseases[J]. Am J Med Genet A, 2012, 158A(11): 2835-42.
[22]Furuya H, Kukita Y, Nagano S, et al. Adult onset globoid cellleukodystrophy (Krabbe disease): analysis of galactosylceramidasecDNA from four Japanese patients[J]. Hum Genet, 1997, 100(3/4):450-6.
[23]Satoh JI, Tokumoto H, Kurohara K, et al. Adult-onset Krabbedisease with homozygous T1853C mutation in the galacto-cerebrosidase gene. Unusual MRI findings of corticospinal tractdemyelination[J]. Neurology, 1997, 49(5): 1392-9.
[24]Hossain MA, Otomo T, Saito S, et al. Late-onset Krabbe disease ispredominant in Japan and its mutant precursor protein undergoesmore effective processing than the infantile-onset form[J]. Gene,2014, 534(2): 144-54.
[25]Zhao S, Zhan X, Wang Y, et al. Large-scale study of clinical andbiochemical characteristics of Chinese patients diagnosed withKrabbe disease[J]. Clin Genet, 2018, 93(2): 248-54.
[26]Zhou X, Yin WW, Yu XF, et al. Adult-onset Krabbe disease due to ahomozygous GALC mutation without abnormal signals on an MRIin a consanguineous family: a case report[J]. Mol Genet GenomicMed, 2020, 8(9): e1407.
[27]Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenasedeficiency[J]. Lancet, 2008, 371(9606): 64-74.
[28]Atay E, Bozaykut A, Ipek IO. Glucose-6-phosphate dehydrogenasedeficiency in neonatal indirect hyperbilirubinemia[J]. J TropPediatr, 2006, 52(1): 56-8.
[29]Katar S. Glucose-6-phosphate dehydrogenase deficiency andkernicterus of South-East Anatolia[J]. J Pediatr Hematol Oncol,2007, 29(5): 284-6.
[30]Mao R, Nelson L, Kates R, et al. Prenatal diagnosis of 21-hydroxylase deficiency caused by gene conversion andrearrangements: pitfalls and molecular diagnostic solutions[J].Prenat Diagn, 2002, 22(13): 1171-6.
[31]Ogino S, Wilson RB. Genetic testing and risk assessment for spinalmuscular atrophy (SMA)[J]. Hum Genet, 2002, 111(6): 477-500.
[32]Talbot K, Ponting CP, Theodosiou AM, et al. Missense mutationclustering in the survival motor neuron gene: a role for a conservedtyrosine and glycine rich region of the protein in RNA metabolism?[J]. Hum Mol Genet, 1997, 6(3): 497-500.
[33]Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidenceand carrier frequency of 5q-linked spinal muscular atrophy-aliterature review[J]. Orphanet J Rare Dis, 2017, 12(1): 124.
[34]Buckley LE, Hopkins MK, Kuller JA. The evolving landscape ofgenetic carrier screening: clinical considerations and challenges[J].Obstet Gynecol Surv, 2023, 78(7): 483-9.
(编辑:经 媛)
基金项目:国家重点研发计划(2021YFC1005303);军队后勤科研项目计划生育专项课题(23JSZ17)
