
基于Nanopore测序技术的非洲猪瘟病毒全基因组测序方法建立
2024-09-19 00:00:00周扬吴炜姿曹伟胜王福广许秀琼钟文霞吴立炀叶健卢受昇
畜牧兽医学报 2024年5期
关键词:生物信息学分析



摘 要: 非洲猪瘟(ASF)是由非洲猪瘟病毒(ASFV)引起的一种高度传染性和致死性疫病,近年来给我国生猪产业的健康发展造成了沉重打击。ASFV庞大的基因组导致人们难以及时掌握流行毒株的全基因组序列。本研究旨在利用Nanopore三代测序技术建立一种简便可靠的ASFV全基因组测序方法。设计覆盖ASFV全基因组的31对引物并分为4个引物池对样本进行扩增,通过Nanopore测序技术对扩增产物进行测序,进一步优化相关生物信息学分析方法,最终成功建立了ASFV 全基因组测序方法。应用该方法成功从某环境拭子样本中获取一株全长为189 416 bp的ASFV全基因组测序。经一代测序验证表明,在B646L、EP402R、E183L、MGF_360-12L、MGF_505-3R和I177L等关键基因及部分变异位点上,本方法结果与一代测序结果一致性100%;在全基因组水平上,本方法结果与二代测序结果一致性为99.94%。此外,在这项研究中,采用Nanopore测序技术发现了NP1450L基因与NP419L基因间区存在56 bp的重复序列插入(通过一代测序技术进行了验证),但是二代测序未能发现这一显著的变异特征。本研究成功建立了基于Nanopore技术的ASFV全基因组测序方法,该方法具有良好的简便性和可靠性,为当前ASF的防控和分子流行病学研究提供了一个重要手段。
关键词: Nanopore测序;非洲猪瘟病毒;生物信息学分析
中图分类号: S852.659.1
文献标志码:A""" 文章编号:0366-6964(2024)05-2080-10
收稿日期:2023-08-10
作者简介:周 扬(1998-),男,广东新兴人,硕士,主要从事动物病原检测技术和三代测序的研究,E-mail: 1418937102@qq.com
*通信作者:卢受昇,主要从事重要动物疫病的防控与研究,E-mail:1179581365@qq.com
A Whole Genome Sequencing Method for African Swine Fever Virus based on Nanopore Sequencing Technology was Established
ZHOU" Yang1,2, WU" Weizi1,2, CAO" Weisheng2, WANG" Fuguang1, XU" Xiuqiong1,
ZHONG" Wenxia1,
WU" Liyang1, YE" Jian1, LU" Shousheng1*
(1.Guangdong Center for Animal Disease Control and Prevention, Guangzhou 510230," China;
2.College of Veterinary Medicine, South China Agricultural University, Guangzhou 510642," China)
Abstract:" African swine fever (ASF) is a highly contagious and deadly disease caused by the African swine fever virus (ASFV), which has dealt a heavy blow to the healthy development of China’s pig industry in recent years. The large genome of ASFV makes it difficult to obtain whole-genome sequence about epidemic strains in a timely manner. This study aims to establish a simple and reliable ASFV whole-genome sequencing method using Nanopore third-generation sequencing technology. Thirty-one primer pairs covering the entire ASFV genome were designed and divided into 4 primer pools to amplify the sample. The amplified product was sequenced by Nanopore sequencing technology, and the relevant bioinformatics analysis methods were further optimized, and finally the ASFV whole-genome sequencing method was successfully established. Whole-genome sequence of ASFV with a total length of 189 416 bp was successfully obtained from an environmental swab sample by this method. Validation through first-generation sequencing has shown that the result of this method is 100% consistent with first-generation sequencing results in key genes and certain variant positions, including B646L, EP402R, E183L, MGF_360-12L, MGF_505-3R, and I177L gene. At the whole-genome level, the consistency between the result of this method and next-generation sequencing(NGS)result is 99.94%. In addition, the utilization of Nanopore sequencing technology in this study revealed a 56 bp repeat sequence insertion within the intergenic region flanked by the NP1450L and NP419L genes. This insertion was subsequently confirmed via first-generation sequencing techniques. Intriguingly, NGS methods failed to detect this distinct variant feature. This study successfully established an ASFV whole-genome sequencing method based on Nanopore technology. This method demonstrates excellent simplicity and reliability, providing an essential tool for the current prevention and control and molecular epidemiological research of ASF.
Key words: nanopore sequencing; African swine fever virus; bioinformatics analysis
*Corresponding author:" LU Shousheng, E-mail: 1179581365@qq.com
非洲猪瘟(African swine fever,ASF)是由非洲猪瘟病毒(African swine fever virus,ASFV)引起的一种猪的急性、热性、高度致死性传染病[1]。2018年8月,我国东北首次报道ASF疫情并成功分离出ASFV[2]。此后,我国多地陆续出现ASF疫情[3-4],大量猪因为感染ASFV而死亡,给我国生猪产业造成了严重的经济损失,严重影响了养猪业的健康发展。ASFV基因组为170~193 kb,包括中央约125 kb的稳定区和左右两侧的可变区[1],左右两侧可变区包含大量MGF基因,这些MGF基因在长期的流行中出现不同模式的变异[5-6]。研究表明,许多基因对ASFV的致病性有重要影响,缺失某些毒力基因的ASFV对猪的致病性明显降低[7-8]。然而,测序技术的局限性和ASFV庞大的基因组导致人们无法及时监测当前流行毒株的全基因组序列。
一代测序又称Sanger测序,其准确性高,被誉为测序技术的“金标准”[9],但其每次测序的有效长度约为800 bp,具有通量低和耗时长的不足,不适用于ASFV的全基因组测序。二代测序(next-generation sequencing,NGS)主要以illumina测序技术和罗氏454测序技术为代表,目前,已广泛应用于病原微生物的全基因组测序[10]。NGS具有高通量和高准确性的优点,但其存在设备昂贵、操作复杂和测序耗时长等不足,不利于疫情的应急溯源。Nanopore测序由英国牛津纳米孔公司(Oxford Nanopore Technologies, ONT)公司开发,属于三代测序(third-generation sequencing,TGS)技术。Nanopore测序的基本原理是将某些生物跨膜蛋白质作为单分子传感器嵌入在高电阻多聚物膜中,通过施加电泳力使带负电荷的生物分子如DNA在DNA解旋酶的作用下解旋并通过跨膜蛋白,期间计算机通过电流变化来捕捉并分析序列信息,从而实现基因组测序。与NGS相比,Nanopore测序具有携带方便、操作便捷、超长读长和单分子实时测序等优势[11]。在人类相关的重大疾病中,Nanopore测序使用已日益成熟。在新型冠状病毒和甲型流感的研究中,基于Nanopore测序平台建立完整的病毒全基因组测序流程并广泛使用[12-13]。当前,猴痘病毒的传播风险日益加深,靶向扩增子结合Nanopore测序的方法已用于猴痘病毒的全基因组测序中,为猴痘病毒的溯源提供了有力的技术支撑[14]。近年来,Nanopore测序在动物病毒的研究中也逐渐兴起。Caserta等[15]基于靶向扩增和Nanopore测序平台首次开发了一套针对猪繁殖与呼吸综合征病毒的全基因组测序程序。此外,在禽流感和口蹄疫等重大动物疫病中也可见Nanopore测序平台应用的报道[16-17]。Nanopore测序的诸多优势使其应用于ASFV全基因组测序的潜力巨大。本研究旨在通过应用Nanopore测序技术,优化相关生物信息学分析方法,建立一种迅速、简便的ASFV全基因组测序方法,为ASF的分子流行病学研究和疫情应急处置溯源提供一种可靠的新手段。
1 材料与方法
1.1 样品来源
样本采自广东地区某农贸市场猪肉档口环境,经亿森宝非洲猪瘟病毒荧光PCR检测试剂盒检测,Ct值为22.62。
1.2 主要试剂
Fast Pure Viral DNA/RNA Mini Kit V2、Fast Pure Gel DNA Extraction Mini Kit购自南京Vazyme公司,Prime STAR GXL DNA Polymerase(R050Q)、Premix Taq DNA Polymerase(RR003Q)购自日本TaKaRa公司,Native Barcoding Expansion(EXP-NBD104)、Flow Cell Wash Kit(EXP-WSH004)、Ligation Sequencing Kit(SQK-LSK110)、Spot-ON Flow Cell(FLO-MIN106D)为英国Oxford Nanopore Technologies公司产品,Qubit 1×dsDNA HS Assay Kit(Q33230)为美国Thermo Fisher Scientific公司产品,AMPure XP beads(A63880)为美国Beckman Coulter公司产品。
1.3 主要仪器
梯度PCR仪为德国Eppendorf公司产品,高速台式冷冻离心机购自湘仪公司,GridION测序仪为英国Oxford Nanopore Technologies产品,磁力架(12321D)、Qubit 4 Fluorometer(Q33226)购自美国Thermo Fisher Scientific公司。
1.4 引物设计
基于我国发现并上传至GenBank的ASFV全基因组序列,使用Oligo 7设计31对引物,引物信息见表1。
1.5 核酸抽提
取200 μL样本,按照Fast Pure Viral DNA/RNA Mini Kit V2试剂盒说明书进行DNA提取。
1.6 靶向扩增子文库构建及GridION测序仪测序
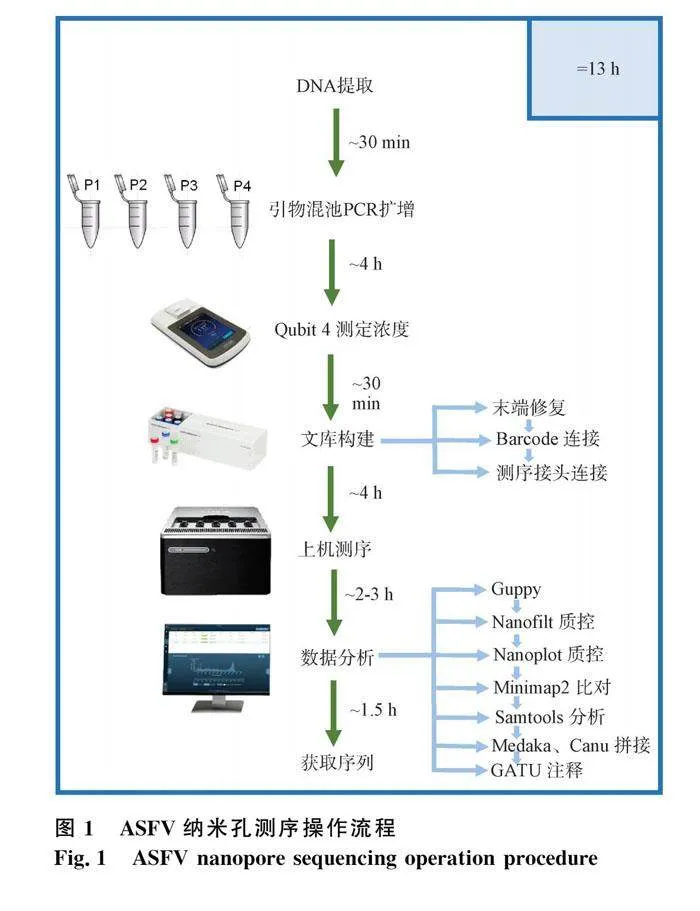
从引物1-F/R~31-F/R中分别取10 μL(10 μmol·L-1)引物,等比混合成4个引物池(Pool),分别为引物池1(pool 1,P1)、引物池2(pool 2,P2)、引物池3(pool 3,P3)和引物池4(pool 4,P4)。其中,P1包含引物1-F/R、3-F/R、5-F/R、7-F/R、9-F/R、11-F/R、13-F/R和15-F/R;P2包含引物2-F/R、4-F/R、6-F/R、8-F/R、10-F/R、12-F/R、14-F/R和16-F/R;P3包含引物17-F/R、19-F/R、21-F/R、23-F/R、25-F/R、27-F/R、29-F/R和31-F/R;P4包含引物18-F/R、20-F/R、22-F/R、24-F/R、26-F/R、28-F/R和30-F/R。将P1~P4作为靶向多重扩增引物,使用Prime STAR GXL DNA Polymerase对样本DNA进行扩增。反应体系为50 μL,包括:5×PrimeSTAR GXL Buffer 10 μL,dNTP Mixture (各2.5 mmol·L-1) 4 μL,引物 (P1、P2、P3或P4) 4 μL (10 μmol·L-1),PrimeSTAR GXL DNA Polymerase 2 μL,模板DNA 5 μL,无菌无核酶水补至50 μL;反应条件:95℃预变性2 min;98℃ 10 s、55℃ 15 s、68 ℃ 4 min,35个循环扩增;68 ℃终延伸5 min。使用AMPure XP磁珠以1∶1的体积比纯化P1~P4的扩增产物并使用Qubit 4荧光计测定浓度(ng·μL-1)。最后从P1~P4分别取600 ng纯化产物,混合成ASFV全基因组扩增子。根据SQK-LSK109建库测序方案进行ASFV文库构建后加入Nanopore Flow cells R9.4测序芯片中,在GridION测序仪上测序。
1.7 生物信息学分析
GridION设备产生的原始数据用经以下软件进行处理分析。测序结果经Guppy转换为fastq格式数据用于后续分析。首先使用NanoFilt(2.8.0)过滤长度小于1 000 bp,质量值低于Q10的测序数据[18];进一步使用Nanoplot(1.41.0)对过滤后的Fstaq数据进行质控,评估reads数、测序质量和读长分布等[18]。以我国代表毒株Pig/HLJ/18(GenBank登录号:MK333180)作为参考基因组,利用Minimap2(2.17-r941)对质控后的测序结果进行比对,并使用SAMtools(1.17)统计每个位点的测序深度及与参考序列匹配的reads数[19],使用Medaka(1.8.1)及Canu完成测序结果拼接,应用GATU进行基因注释[20](图1)。
1.8 测序结果准确性验证
1.8.1 NGS验证
为验证Nanopore测序的准确性,将P1~P4的扩增产物交由广东美格基因科技有限公司进行NGS。通过MAFFT软件对NGS结果和Nanopore测序结果进行比对以验证Nnaopore测序的准确性。
1.8.2 一代测序验证
使用Oligo 7对ASFV的B646L、EP402R、E183L、I177L、MGF_360-12L和MGF_505-3R等关键基因设计引物进行扩增。将Nanopore测序结果与Pig/HLJ/18进行比对,随机选取部分变异位点,进一步设计引物进行扩增测序,验证变异位点的准确性。一代测序由生工生物工程(上海)股份有限公司完成。
2 结 果
2.1 PCR扩增
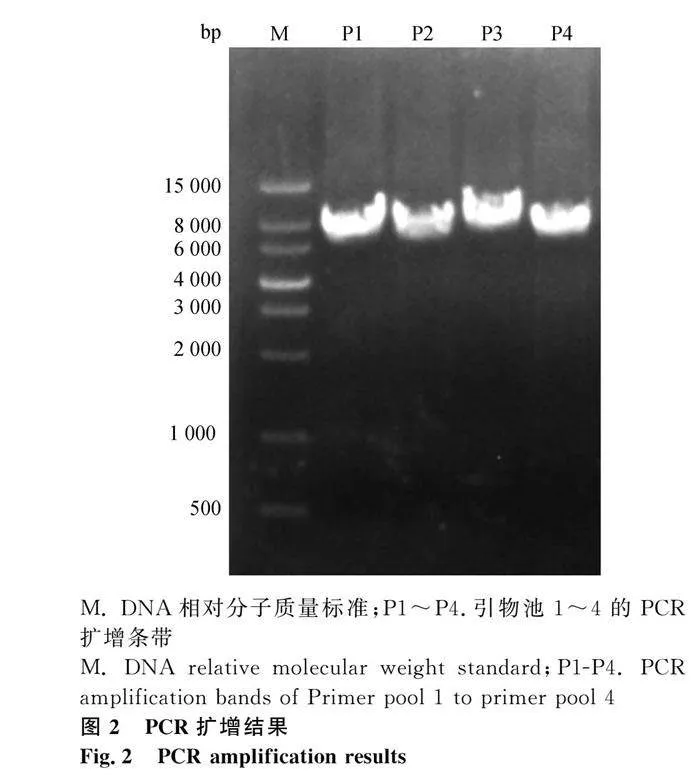
将引物等比混合成P1~P4后进行靶向多重PCR扩增,取5 μL PCR产物用于琼脂糖凝胶电泳,结果表明各引物池均扩增出预期条带(图2)。
2.2 生物信息学分析
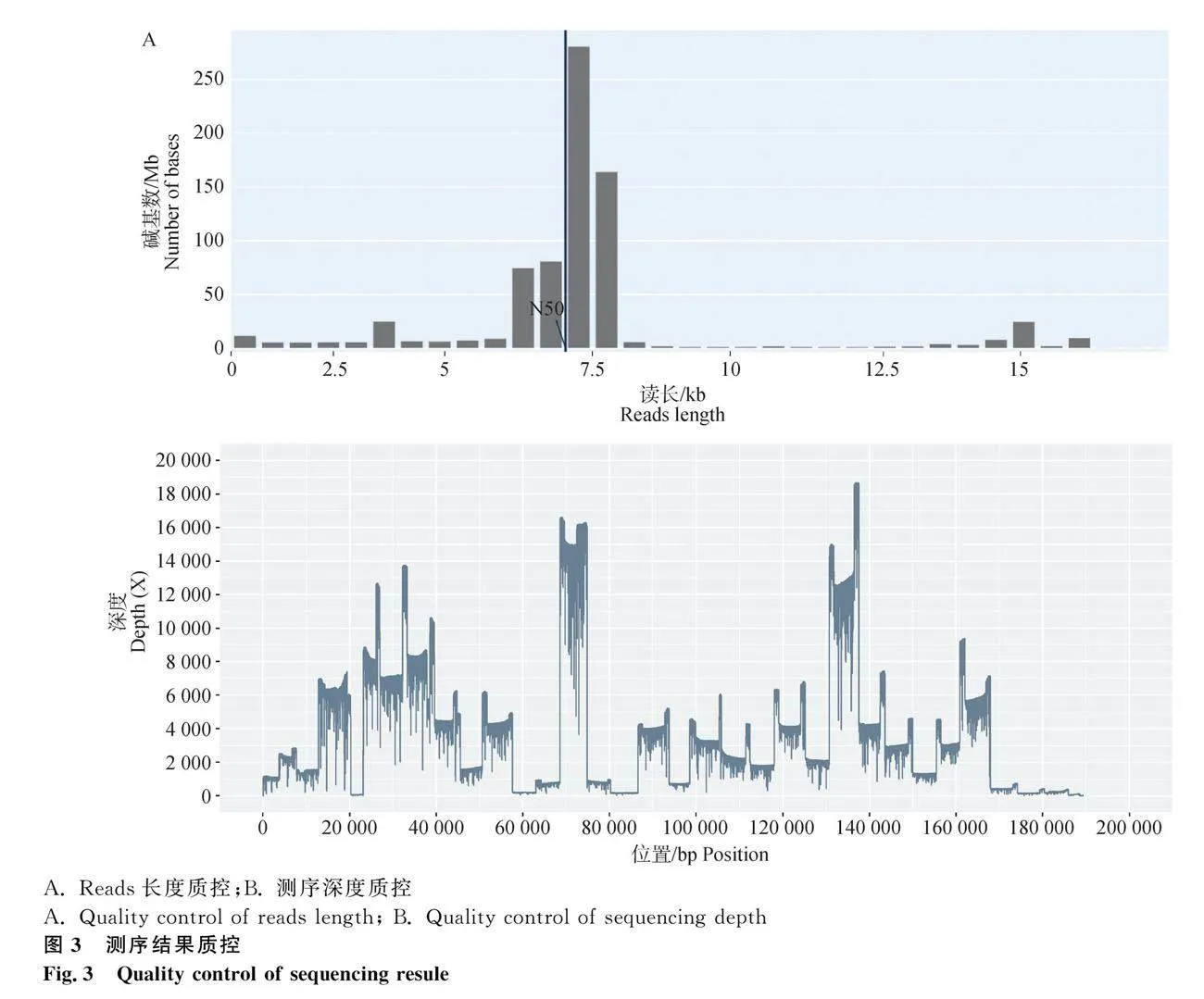
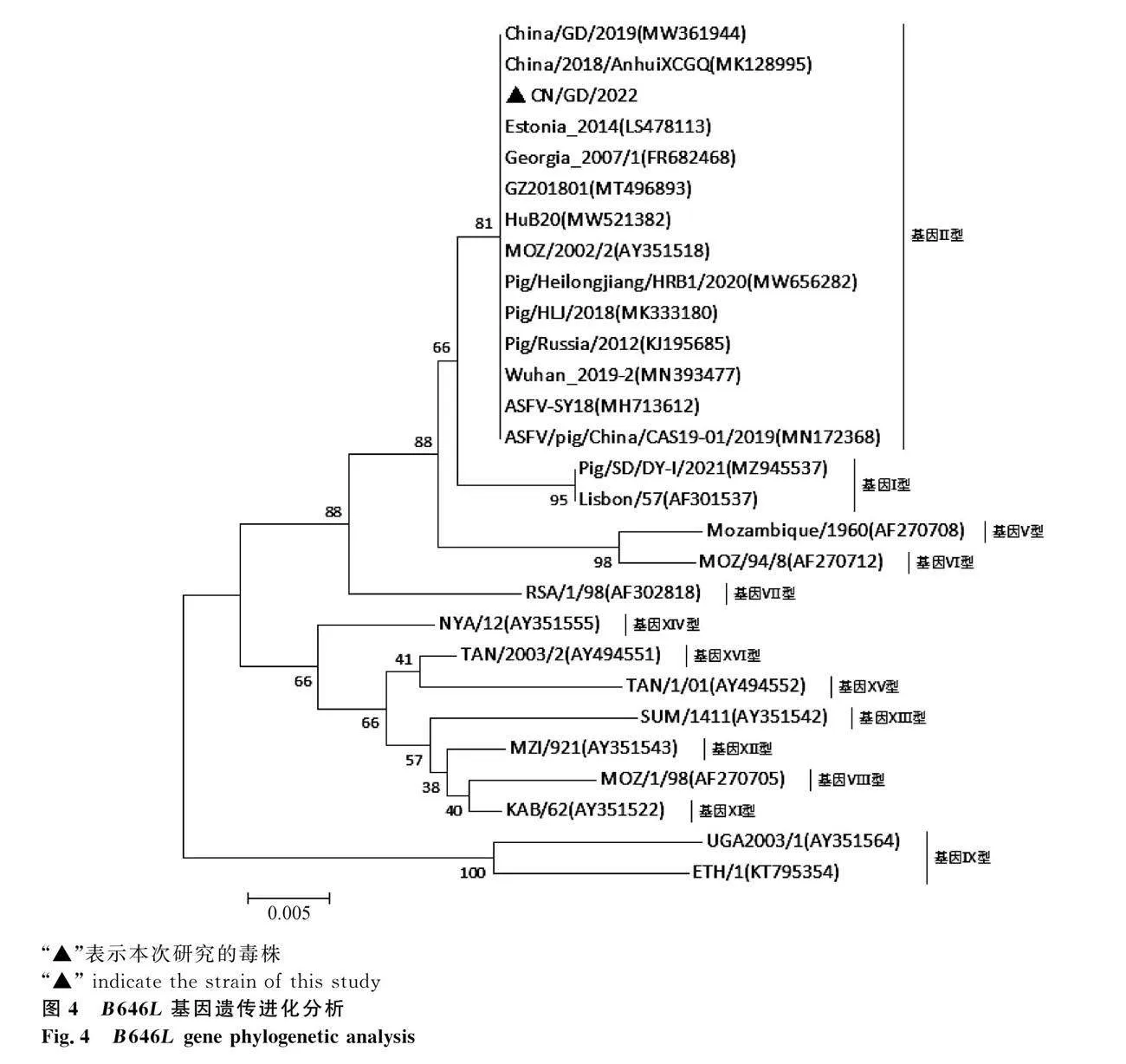
经测序成功获得1.51 G的Fastq原始数据,使用Nanofilt软件过滤长度低于1 000 bp及测序质量低于Q10的测序数据后,余1.47 G Fsatq原始数据。经Nanoplot软件质控表明过滤后reads数为124 126条,平均每条reads长度为6 174.2 bp,reads长度N50为7 096 bp(图3A)。过滤后碱基的准确性gt;90%(Q10),平均reads质量为Q13。SAMtools软件分析表明有98.04%的reads与参考序列匹配(表2)。测序深度统计表明本次测序的最大测序深度为18 684×,平均测序深度为3 906×(图3B),其中测序深度大于50×的碱基位点占比为99.53%,但存在覆盖度不均匀的现象。使用Medaka和canu软件完成序列拼接,得到长度为189 416 bp的样本全基因组序列(CN/GD/2022,GenBank登录号:OR290104),GATU注释表明该毒株包含183个ORF。通过ASFV B646L 基因3′端部分序列遗传进化分析表明CN/GD/2022属于基因Ⅱ型ASFV(图4)。
2.3 Nanopore测序结果准确性验证
2.3.1 NGS验证
广东美格基因科技有限公司最终返回全长为188 491 bp的ASFV NGS结果,经MAFFT全基因组比对表明NGS结果与本方法测序结果一致性为99.94%。在B646L、EP402R、E183L、MGF_505-3R、MGF_360-12L和I177L等关键基因上,Nanopore测序和NGS结果完全一致;随机选取6处变异位点验证,在其中5处变异位点中NGS结果和Nanopore测序结果相同(表3)。同时,基于参考毒株Pig/HLJ/18,本次验证发现NGS结在MGF_505-9R与MGF_505-10R基因间非编码区测出2次“TCAATAGTTTAGTTAAG”的重复序列,而Nanopore测序测出3次重复序列(图5A);进一步比对发现在NP1450L和NP419L基因间区,NGS结果与Nanopore测序结果差异较大,NGS结果表明该处存在56 bp序列插入,而NGS结果与参考序列一致(图5 B)。
2.3.2 一代测序验证
通过MEGA 7比对表明,在关键基因如B646L、EP402R、E183L、MGF_505-3R、MGF_360-12L和I177L基因上,Nanopore测序结果与一代测序结果一致性为100%;在6处变异位点上,Nanopore测序在其中5处变异位点上的测序结果与一代测序结果相符(表3)。本次试验发现在重复序列上Nanopore测序结果与一代测序结果一致,实现MGF_505-9R与MGF_505-10R基因间非编码区中3次“TCAATAGTTTAGTTAAG”的重复序列的测序(图5 A)。进一步验证发现NP1450L和NP419L基因间区存在56 bp的小片段插入(GenBank登录号:OR290103),与NP419L基因中部分序列形成连续重复序列(图5 B),Nanopore测序成功发现该变异特征。
3 讨 论
自2018年ASFV入侵我国以来,非洲猪瘟在我国广泛传播并趋于稳定。然而,ASFV的基因组极为复杂,其中许多基因功能至今尚不明确,且迄今尚未开发出有效的疫苗来防控ASFV的传播。随着非洲猪瘟在我国的传播时间延长,从早期报道的缺失株到近期报道的重组毒株,ASFV流行情况变得更加复杂和多样化[21]。
近年来,Nanopore测序长读长及高产量的优势,使其逐渐应用于具有较大基因组的病原体的测序中,包括猴痘病毒、埃博拉病毒、多种细菌和真菌等[22-24]。通过预训练算法,Nanopore测序技术可以通过未修饰碱基和修饰碱基的纳米孔读数产生的电流强度来检测DNA修饰,用于表观遗传学的研究[25]。与NGS相比,Nanopore测序设备具有简化的操作流程、轻便的设备、以及较低的能耗。以Goordial等[26]的研究为例,他们成功将Nanopore测序设备运往北极地区,对土壤样本进行了宏基因组测序,从而揭示了冻土中微生物群落的关键信息。另外,Castro-Wallace等[27]的研究表明,将Nanopore测序平台运送至国际空间站后,通过对预先设置的样本进行测序和分析,证实了Nanopore测序平台在国际空间站上复杂的环境中能够实现与地面相当的性能水平。本次试验采用Nanopore连接法进行ASFV文库构建,从样本核酸提取到完成测序时长约为13 h;试验过程中主要使用的设备有PCR仪、GridION测序仪和分析服务器。Nanopore测序不仅大大减少了试验工作量,而且在资源有限的情况下使用更加方便。以上优势使Nanopore测序在疫情暴发初期及时监测毒株的全基因组序列方面具有良好的潜力。
测序深度、错误率和reads长度等指标对测序结果的准确性有直接的影响。本次试验的平均测序深度达到了3 905×,而reads的长度N50值为7 096 bp。长reads不仅可以减少组装和分析测序结果的复杂性,而且更好检测到某些重复序列和结构变异。然而,通过测序深度分析发现本次试验在不同基因区域的测序深度存在差异,这可能会导致一些覆盖率低的基因位点的准确性降低。这种现象与靶向多重PCR反应中各引物的扩增效率不一致密切相关。在后续的试验中,可以优化引物池中各引物的比例,以促进扩增片段在ASFV的基因组中均匀分布。Jia等[3]使用Nanopore测序技术对ASFV进行全基因组测序的研究结果显示,下机数据中仅包含0.15%的ASFV reads,平均测序深度为330.08×。相比之下,本次试验的结果更为可靠。通过采用靶向扩增技术,本试验中ASFV reads占据了样本下机数据的98.04%,平均测序深度达到了3 906×。因此,这种方法能够富集样本中的病毒核酸,减少宿主基因比例,从而通过提高测序深度来提高测序结果的准确性。此外,Nanopore测序的准确性受到纳米孔蛋白活性、化学建库试剂和数据分析方法等多个因素的影响。NGS可保证99%以上的准确性,相对而言,Nanopore测序的准确性较低,通常在85%~99%。在本次试验中,Nanopore测序的结果与NGS的一致性达到了99%以上,而且经过一代测序验证了部分变异位点的准确性。随着芯片的更迭以及碱基调用算法的不断改进,Nanopore测序的准确性将会不断提高。Nanopore测序依赖于通过纳米孔的DNA分子产生的电流信号来识别和记录碱基序列。不同的碱基会产生不同的电流信号,因此可以根据电流信号来识别相应的碱基。当一连串的相同碱基通过纳米孔时,电流信号可能变得相似,难以准确区分,这是导致本研究中Nanopore测序在连续的A、T、C或G区域表现出较低的准确性的主要原因。但在最新的研究中,通过优化纳米孔蛋白材料、碱基调用算法和测序结果质控纠错程序,Nanopore测序的准确性在不断提高[28]。DNA中的重复序列在各种不同的生物种类中都广泛存在,如近一半的人类基因组被重复序列覆盖。同样,ASFV庞大的基因组中存在一定量的串联重复序列[29]。经一代测序、NGS及Nanopore测序结果比对,本研究发现NGS未能正确实现ASFV MGF_505-9R和MGF_505-10R基因间区及NP1450L和NP419L基因间区的重复序列的测序。一方面,NGS读长短(~250 bp),容易在测序结果的比对和组装过程中产生歧义,无法准确识别出重复序列[30];另一方面,Nanopore测序为单分子长读长测序,理论上通过纳米孔的单条DNA序列有多长即可测多长,该优势使其在实现重复序列和结构变异等复杂基因组的测序上更具优势。因此,在ASFV的全基因组测序中,可以将Nanopore测序与NGS相结合,以获取高质量的流行毒株全基因组。
当前我国除了基因Ⅱ型ASF流行之外,还陆续报告了基因Ⅰ型、基因缺失型和重组型ASFV。在这种情况下,可以根据基因Ⅰ型和基因Ⅱ型流行毒株的共同保守位点设计引物,并单独设计跨越常见MGF基因缺失区域的引物,将它们合并为一个引物池。这样的策略可以实现临床样本的靶向扩增并应用于Nanopore测序,以应对我国当前复杂多变的ASFV流行情况。经B646L基因C端部分序列可将ASFV分为24个基因型[31],经遗传进化分析表明CN/GD/2022为基因Ⅱ型ASFV,属于目前我国主要流行的基因型。因此,本次建立的Nanopore测序平台适用于我国流行ASFV的全基因组测序。
4 结 论
本研究基于Nanopore测序平台成功建立了ASFV 全基因组测序方法,通过该方法成功实现了临床ASFV阳性样本的全基因组测序。经分析表明,该方法具有快速、准确、操作简便等优点,为ASF诊断、监测和预警提供了一个重要的手段。
参考文献(References):
[1] ALONSO C, BORCA M, DIXON L, et al. ICTV virus taxonomy profile:Asfarviridae[J]. J Gen Virol, 2018, 99(5):613-614.
[2] ZHAO D M, LIU R Q, ZHANG X F, et al. Replication and virulence in pigs of the first African swine fever virus isolated in China[J]. Emerg Microbes Infect, 2019, 8(1):438-447.
[3] JIA L J, JIANG M W, WU K, et al. Nanopore sequencing of African swine fever virus[J]. Sci China Life Sci, 2020, 63(1):160-164.
[4] 张艳艳, 张静远, 杨金金, 等. 1株非洲猪瘟病毒自然变异毒株的鉴定[J]. 中国兽医学报, 2021, 41(2):199-207.
ZHANG Y Y, ZHANG J Y, YANG J J, et al. Identification of a natural variant of African swine fever virus in China[J]. Chinese Journal of Veterinary Science, 2021, 41(2):199-207.
[5] SHI K C, LIU H X, YIN Y W, et al. Molecular characterization of African swine fever virus from 2019-2020 outbreaks in Guangxi Province, Southern China[J]. Front Vet Sci, 2022, 9:912224.
[6] SUN Y K, XU Z Y, GAO H, et al. Detection of a novel African swine fever virus with three large-fragment deletions in genome, China[J]. Microbiol Spectr, 2022, 10(5):e0215522.
[7] TEKLUE T, WANG T, LUO Y Z, et al. Generation and evaluation of an African swine fever virus mutant with deletion of the CD2v and UK genes[J]. Vaccines (Basel), 2020, 8(4):763.
[8] CHEN W Y, ZHAO D M, HE X J, et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs[J]. Sci China Life Sci, 2020, 63(5):623-634.
[9] SANGER F, NICKLEN S, COULSON A R. DNA sequencing with chain-terminating inhibitors[J]. Proc Natl Acad Sci U S A, 1977, 74(12):5463-5467.
[10] MARDIS E R. Next-generation DNA sequencing methods[J]. Annu Rev Genom Hum Genet, 2008, 9(1):387-402.
[11] LIN B, HUI J A, MAO H J. Nanopore technology and its applications in gene sequencing[J]. Biosensors (Basel), 2021, 11(7):214.
[12] KING J, HARDER T, BEER M, et al. Rapid multiplex MinION nanopore sequencing workflow for influenza A viruses[J]. BMC Infect Dis, 2020, 20(1):648.
[13] LAMBISIA A W, MOHAMMED K S, MAKORI T O, et al. Optimization of the SARS-CoV-2 ARTIC network V4 primers and whole genome sequencing protocol[J]. Front Med (Lausanne), 2022, 9:836728.
[14] LAITON-DONATO K, LVAREZ-DAZ D A, FRANCO-MUOZ C, et al. Monkeypox virus genome sequence from an imported human case in Colombia[J]. Biomedica, 2022, 42(3):541-545.
[15] CASERTA L C, ZHANG J Q, PIEYRO P, et al. Rapid genotyping of porcine reproductive and respiratory syndrome virus (PRRSV) using MinION nanopore sequencing[J]. PLoS One, 2023, 18(5):e0282767.
[16] DE VRIES E M, COGAN N O I, GUBALA A J, et al. Rapid, in-field deployable, avian influenza virus haemagglutinin characterisation tool using MinION technology[J]. Sci Rep, 2022, 12(1):11886.
[17] BROWN E, FREIMANIS G, SHAW A E, et al. Characterising foot-and-mouth disease virus in clinical samples using nanopore sequencing[J]. Front Vet Sci, 2021, 8:656256.
[18] DE COSTER W, D’HERT S, SCHULTZ D T, et al. NanoPack: visualizing and processing long-read sequencing data[J]. Bioinformatics, 2018, 34(15):2666-2669.
[19] DANECEK P, BONFIELD J K, LIDDLE J, et al. Twelve years of SAMtools and BCFtools[J]. Gigascience, 2021, 10(2):giab008.
[20] TCHEREPANOV V, EHLERS A, UPTON C. Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome[J]. BMC Genomics, 2006, 7:150.
[21] ZHAO D M, SUN E C, HUANG L Y, et al. Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs[J]. Nat Commun, 2023, 14(1):3096.
[22] QUICK J, LOMAN N J, DURAFFOUR S, et al. Real-time, portable genome sequencing for Ebola surveillance[J]. Nature, 2016, 530(7589):228-232.
[23] PETERSEN L M, MARTIN I W, MOSCHETTI W E, et al. Third-generation sequencing in the clinical laboratory: exploring the advantages and challenges of nanopore sequencing[J]. J Clin Microbiol, 2019, 58(1):e01315-19.
[24] ASHIKAWA S, TARUMOTO N, IMAI K, et al. Rapid identification of pathogens from positive blood culture bottles with the MinION nanopore sequencer[J]. J Med Microbiol, 2018, 67(11):1589-1595.
[25] LIU Y, ROSIKIEWICZ W, PAN Z W, et al. DNA methylation-calling tools for Oxford Nanopore sequencing: a survey and human epigenome-wide evaluation[J]. Genome Biol, 2021, 22(1):295.
[26] GOORDIAL J, ALTSHULER I, HINDSON K, et al. In situ field sequencing and life detection in remote (79°26′N) Canadian high arctic permafrost ice wedge microbial communities[J]. Front Microbiol, 2017, 8:2594.
[27] CASTRO-WALLACE S L, CHIU C Y, JOHN K K, et al. Nanopore DNA sequencing and genome assembly on the international space station[J]. Sci Rep, 2017, 7(1):18022.
[28] WANG Y H, ZHAO Y, BOLLAS A, et al. Nanopore sequencing technology, bioinformatics and applications[J]. Nat Biotechnol, 2021, 39(11):1348-1365.
[29] KIM S H, LEE S I, JEONG H G, et al. Rapid emergence of African swine fever virus variants with different numbers of a tandem repeat sequence in South Korea[J]. Transbound Emerg Dis, 2021, 68(4):1726-1730.
[30] TREANGEN T J, SALZBERG S L. Repetitive DNA and next-generation sequencing: computational challenges and solutions[J]. Nat Rev Genet, 2011, 13(1):36-46.
[31] ACHENBACH J E, GALLARDO C, NIETO-PELEGRN E, et al. Identification of a new genotype of African swine fever virus in domestic pigs from ethiopia[J]. Transbound Emerg Dis, 2017, 64(5):1393-1404.
(编辑 白永平)
猜你喜欢
中国中药杂志(2017年2期)2017-03-25 17:25:49
湖北农业科学(2017年3期)2017-03-21 19:18:04
中国中药杂志(2017年1期)2017-03-06 21:25:43
江苏农业科学(2016年6期)2016-07-25 18:03:06
江苏农业科学(2016年5期)2016-07-23 01:23:23
江苏农业科学(2016年4期)2016-06-14 01:50:00
江苏农业科学(2016年3期)2016-05-03 20:03:17
江苏农业科学(2016年2期)2016-04-11 20:17:35
江苏农业科学(2015年11期)2016-01-27 14:28:11
江苏农业科学(2015年1期)2015-04-17 20:52:43
