
p53V149F 突变打靶载体及细胞模型的构建
2024-08-04 00:00:00曹攀蔡洁张晓银熊喆角德灵赵恒徐凯祥赵红业
云南农业大学学报(自然科学) 2024年3期



摘要: 【目的】构建p53 基因突变模型,为研究p53 基因突变引发肿瘤的发生和进展提供细胞模型资源。【方法】利用CRISPR/Cas9 基因编辑技术,根据 p53V149F 突变位点的序列,在线设计合成单链向导sgRNA,构建CRISPR 打靶载体;将重组载体转染细胞,流式分选阳性细胞群,提取细胞基因组,Sanger 测序分析Cas9 核酸酶对靶位点的切割情况,构建同源修复模板,通过T7EN1 试验进一步检测编辑效率;将重组载体和同源模板通过电转染方法共同转染至成纤维细胞中,通过单克隆挑取、PCR 和Sanger 测序鉴定挑取含目标突变的细胞株。【结果】成功构建靶向p53 基因目标区域的CRISPR/Cas9 打靶载体;获得5 个含目标突变的克隆点,点突变率为10%,测序结果出现套峰,表明出现基因型多样化;1 个细胞克隆的测序结果无杂峰,TA 克隆鉴定结果表明:一个等位基因存在p53V149F 位点突变,另一个等位基因存在240 bp 缺失。【结论】成功构建了含p53V149F 的细胞系,并对基因的功能进行了初步验证,为进一步构建两基因同时修饰模型奠定了基础,并为药物筛选提供可用的细胞模型资源,为创制模拟疾病发生的遗传模式和临床表现的小型猪实验动物模型奠定了基础。
关键词: 基因编辑;打靶载体;癌症;p53 基因
中图分类号: Q78 文献标志码: A 文章编号: 1004–390X (2024) 03−0064−08
CRISPR/Cas9 技术是构建基因编辑模型的重要手段,其优势使其成为生物和治疗应用基因工程的有利工具,被用于在活细胞或生物体中抑制/激活基因表达或标记特定基因组位点的功能域,以探索发育机制、基因表达调控和动物行为[1]。CRISPR/Cas9 系统作为通过靶向基因修饰进行作物改良的有力工具,可对基因组序列进行精确地切割和修饰。ZHAO 等[2]利用CRIPSRi 系统在猪的细胞水平抑制了导入的GFP 基因表达,为在哺乳动物中开展基因表达调控研究提供了良好的借鉴。在研制用于器官移植和其他疾病研究的家猪模型中,已有研究利用CRISPR/Cas9 技术获得猪肾细胞系PK15 中全部62 个拷贝的 PERV pol(多聚酶) 基因敲除的猪肾细胞系[3]。此外,在动物疾病模型的研究中,已获得了皮肤白化病、帕金森疾病相关基因敲除体细胞克隆猪[4]以及白化病症状的兔子[5]。CRISPR/Cas9 基因编辑技术旨在使精确的靶点产生目的基因突变,从而获得目的靶向基因区域的打靶载体,通过有效的技术解析肿瘤发生的机制,确定药物开发的靶点,提高武装细胞进行治疗的可能性,进而加速癌症研究[6]。
从20 世纪被发现以来,癌症的发病率不断上升,全球每年登记的癌症新病例超过1 400 万例[7],其特征是异常细胞不受控制地增殖和免疫系统的异常识别,被认为是“永不愈合的伤口”,但若能及早发现,所有的癌症都可以治愈[8]。癌症的常规治疗主要靠消灭癌细胞或抑制其增殖,但复发的患者依然占比较高,无法获得长期生存。因此,充分了解癌症从发病到转移再到复发的机制对其治疗具有重要意义。近年来,国内外学者对癌症的发病机制进行了多方面的研究,结果表明:癌症的发病与p53、K-Ras、EGFR、P16等多个基因突变相关,特别是与p53 显著相关[9]。约50% 的人类癌症中存在p53 突变[10],在某些特定癌症的亚型中,如三阴性乳腺癌[11]、转移性微卫星阳性的结直肠癌[12]、鳞状食管癌[13]等,至少80% 的病例发生p53 突变。由于p53 参与了多种肿瘤抑制途径,其功能在人类癌症中经常受损。p53 突变通常是功能丧失,也可能提供新形态(功能获得) 活动,如促进癌细胞干性、细胞增殖、侵袭和转移,从而促进癌症发展[14]。
p53 作为一种关键的肿瘤抑制因子[15],受细胞内外众多的信号刺激,通过抑制细胞生长、诱导细胞凋亡、促进DNA 损伤修复等过程抑制肿瘤的发生和发展。在应对DNA 损伤和异常生长信号时, p53 和鼠双微体(murine double minute2,MDM2) 基因之间的相互作用被阻断,p53变得稳定,使得p53 主要通过靶基因的转激活来调节一系列不同的细胞反应。持续的DNA 损伤会阻止转录和复制,从而阻碍细胞功能并促进细胞衰老和凋亡。DNA 损伤、电离辐射和活性氧都可以稳定突变的p53[16-17],突变主要发生在125~300 密码子的DNA 结合区域内[18],这是p53 与DNA 结合发挥转录因子功能的必需区域。p53在恶性肿瘤中几乎普遍失活,因此,它是抗癌新药开发中极具吸引力的靶点。p53 激活的结果受其动态、与其他蛋白质的相互作用以及翻译后修饰的控制。尽管已有多种针对功能失调的p53 治疗癌症的策略,但到目前为止,只有2 种策略产生了用于临床试验的化合物[19]。本研究以猪成纤维细胞构建含p53V149F 的细胞系,并对基因功能进行初步验证,以期为进一步构建两基因同时修饰的模型奠定基础,并为药物筛选提供可用的细胞模型资源,也为创制模拟疾病发生的遗传模式和临床表现的小型猪实验动物模型奠定基础。
1 材料与方法
1.1 试验材料
供试质粒:PGL3-U6-sgRNA-EGFP 和spCas9,由上海科技大学黄行许教授课题组馈赠;供试33 日龄滇南小耳猪成纤维细胞系由云南省动物基因编辑与体细胞克隆技术重点实验室提供。
1.2 p53sgRNA 载体设计与构建
通过网站http://crispr.mit.edu 设计与人157 密码子区域同源的猪p53 基因(NC_010454.4) V149第5 号外显子的sgRNA (TGGCACCCGTGTCCGCGCCA)。以滇南小耳猪DNA 为模板,以p53-snp(F:CAGCTATGATTTCCGTCTAGGG; R:GCTGTGTCGAAAAGTGTTTCTG)为引物进行PCR扩增,扩增片段为400 bp。反应程序为:95 ℃,5 min;95 ℃,30 s;68~58 ℃ (每进行1 个循环退火温度下降1 ℃),30 s;72 ℃,1 min;10 个循环;95 ℃,30 s;58 ℃,30 s;72 ℃,1 min;25 个循环;72 ℃,7 min;12 ℃ 保存。PCR 结束后对产物进行凝胶电泳,将PCR 产物进行Sanger 测序,比对分析峰图并进行sgRNA 的SNP 检测;将sgRNA 正反链退火后形成双链,连接到线性化的载体PGL3-U6-sgRNA-EGFP 和PGL3-U6-sgRNA-puro,连接产物转化涂板,利用氨苄青霉素抗性LB 平板筛选,挑取菌落进行PCR 鉴定,并将其中1 个阳性克隆菌进行Sanger 测序,以获得正确的PGL3-U6-p53sgRNA 载体,再通过试剂盒(北京天根,DP118-02) 提取PGL3-U6-p53sgRNA和spCas9 质粒备用。
1.3 细胞培养
滇南小耳猪成纤维细胞系培养于含有10% 胎牛血清和1% 双抗的DMEM 培养液,置于37 ℃培养箱中(5% CO2),待细胞的生长覆盖率达到60%~70% 时收集细胞用于后续转染试验。
1.4 p53sgRNA 活性鉴定和供体构建
将提取的质粒PGL3-U6-p53sgRNA-EGFP 和spCas9 按照1∶2 的比例通过电转染(LONZA4D-Nucleofector) 猪成纤维细胞(即试验组),以只加入转染试剂、不加质粒的处理为对照组,48 h后于荧光显微镜下观察绿色荧光。采用流式细胞分选仪分选绿色荧光阳性细胞,收集阳性细胞,离心去除上清液,加细胞裂解液10 μL 裂解细胞,取样品1 μL 为模板进行PCR 扩增,程序、引物和体系同1.2 节。将PCR 产物送至测序公司进行Sanger 测序,利用Snapgene 软件进行序列比对,分析sgRNA 对p53 基因的编辑情况;通过T7EN1 对PCR 纯化产物进行酶切,程序为:95 ℃,5 min;94 ℃,2 s,200 个循环(每进行1 个循环退火温度下降0.1 ℃);75 ℃,600 个循环(每进行1 个循环退火温度下降0.1 ℃);16 ℃,2 min,进行退火反应。反应结束后向体系中加入T7EN1 酶0.5 μL,置于恒温水浴锅中37 ℃ 酶切30 min;加入5×Loading Buffer 灭活酶,使用1.2% 凝胶进行电泳鉴定;将sgRNA 打靶位置的左右50 nt 基因序列(包含C 到T 突变碱基) 进行ssODN 合成作为供体。
1.5 p53V149F 细胞系的单克隆培养与鉴定
将提取的质粒PGL3-U6-p53sgRNA-puro、质粒spCas9、ssODN 按照1∶2∶2 的比例通过电转染猪成纤维细胞48 h,利用有限稀释的方法分离和培养单克隆细胞。具体为:加入3 μg/mL 嘌呤霉素筛选阳性细胞,48 h 后收集细胞;按照每皿100~150 个的密度接种到培养皿(直径9 cm) 中,单克隆生长培养15 d,取单细胞克隆点鉴定基因型;以裂解液裂解的单细胞克隆点作为模板,通过PCR 和Sanger 测序鉴定p53 基因位点的编辑情况,程序、引物和体系同1.2 节;再将克隆点PCR 产物进行TA 克隆,通过Sanger 测序分析基因型。
2 结果与分析
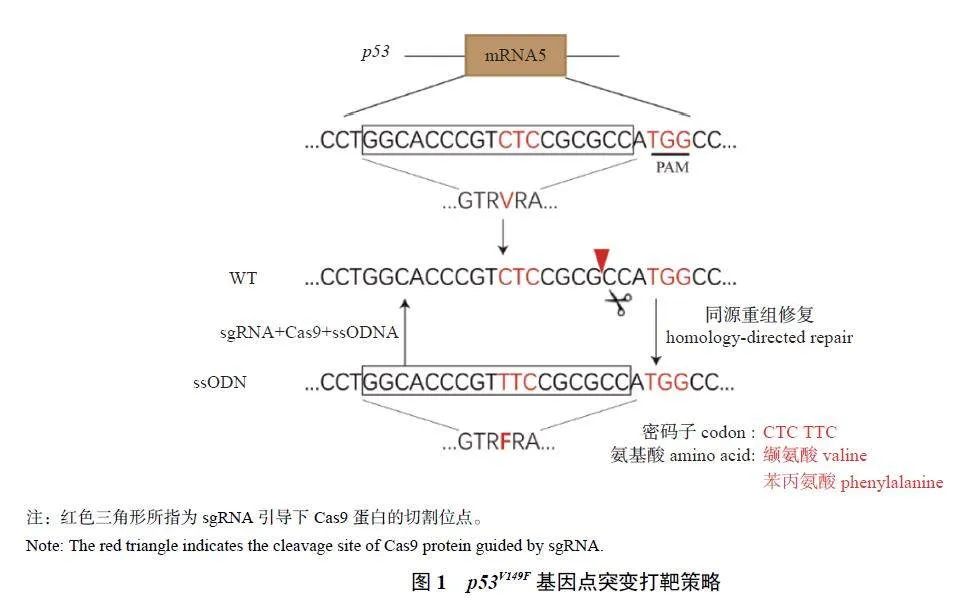
2.1 内源p53 基因打靶策略
猪p53 基因mRNA 总长1 849 bp,编码386个氨基酸。在猪p53 基因第5 号外显子第149 号氨基酸编码的碱基位置设计sgRNA (图1),连接到sgRNA 的表达载体骨架,根据细胞水平的活性验证找到Cas9 的切口位置(PAM 结构上游3、4 号碱基之间),设计包含目标突变的ssODN。通过共转染sgRNA 质粒、Cas9 质粒和ssODN,对目标区域进行切割,造成双链断裂。由于提供了外源重组模板,通过机体同源重组修复机制从而引入点突变,实现p53 基因的第149 号氨基酸由缬氨酸突变为苯丙氨酸,对应p53 基因mRNA5的94 号碱基由胞嘧啶(C) 突变为鸟嘌呤(G),从而获得p53V149F。
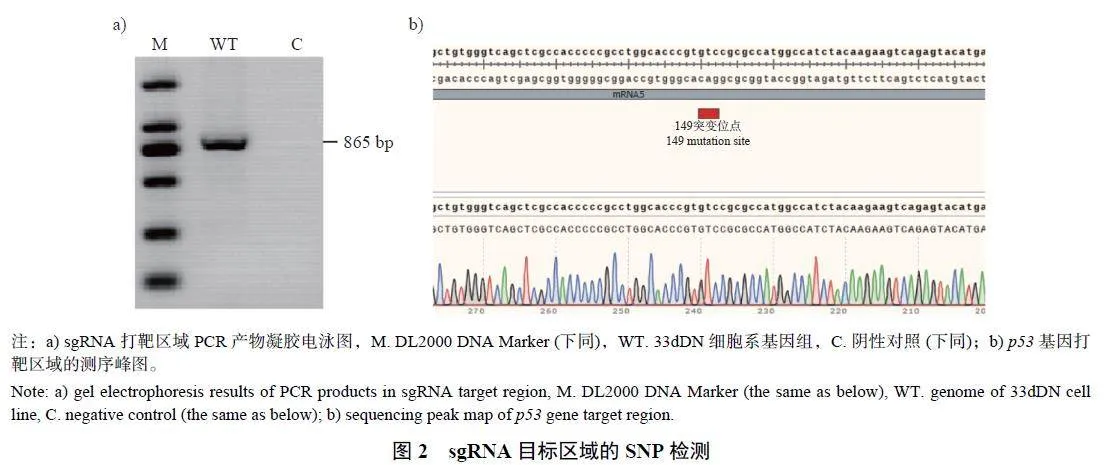
2.2 p53sgRNA 目标区域SNP 检测
滇南小耳猪成纤维细胞系DNA 通过PCR 扩增和凝胶电泳,获得865 bp 的目的条带(图2a)。将PCR 产物进行Sanger 测序比对基因组序列,结果显示:峰图无杂峰且与参考基因组序列一致(图2b)。表明猪p53 的sgRNA 打靶区域不存在SNP 位点,可直接用于sgRNA 打靶设计。
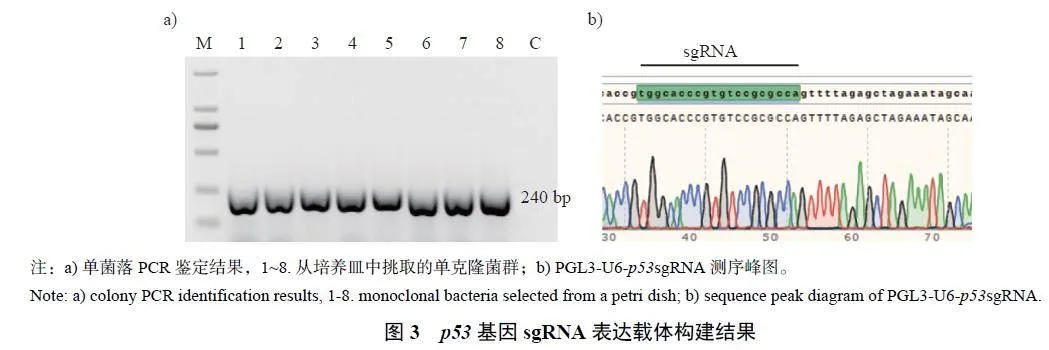
2.3 p53 基因sgRNA 表达载体构建结果
经氨苄青霉素抗性LB 平板筛选,共挑取8个单菌落进行PCR 鉴定,PCR 产物凝胶电泳结果显示:8 个菌落都能扩增出单一的240 bp 目的条带,与目标大小一致(图3a);阳性克隆菌的Sanger测序结果表明已成功构建PGL3-U6-p53sgRNApuro和PGL3-U6-p53sgRNA-EGFP 载体(图3b)。
2.4 p53 基因sgRNA 表达载体细胞水平验证
质粒PGL3-U6-sgRNA-EGFP 和spCas9 电转染进入细胞48 h 后,观察到较强的GFP 荧光蛋白表达,而对照组未观察到荧光,表明p53 基因sgRNA 表达载体成功转染进细胞(图4a);T7EN1酶切试验结果显示:野生型原始条带(865 bp) 未被切割,而共转染PGL3-U6-p53sgRNA-EGFP 和spCas9 质粒的细胞基因组PCR 条带被切割为597和268 bp 的目的条带(图4b),表明在sgRNA 打靶位置Cas9 核酸酶发生了靶向切割;Sanger 测序峰图显示:从PAM 前3、4 号碱基之间出现杂峰(图4c) 。
2.5 p53 基因供体构建结果
选择切口位置(目标区域sgRNA 的PAM 结构上游3、4 号碱基之间) 左右各50 nt 作为同源臂,合成含C 到T 突变位点的ssODN 作为同源重组修复的模板(图5)。
2.6 单克隆细胞筛选及鉴定
经过PCR 扩增和Sanger 测序50 个克隆点中,共有5 个克隆点含有目标点突变(图6a),其中P36 号克隆点测序未出现杂峰(图6b);该克隆点的TA 克隆和Sanger 测序鉴定结果显示:该克隆点为单等位基因V149F 位点突变,缺失240 bp片段(图6c)。
3 讨论
对p53 基因的编码区进行序列检测发现:86% 的突变聚集在125~300 密码子[20],而目前针对p53 突变的癌症模型构建以R175H (猪167 位点同源) 为主[21-23]。有研究绘制了加合物在人类p53 基因的分布, 在密码子157 (猪149 同源)CpG 序列的鸟嘌呤上形成了强且有选择性的加合物,这是导致肺癌发生的主要突变位点[24],但目前鲜有对于该位点的研究报道。本研究获得一个存在p53V149F 的p53 等位基因;而另一个等位基因存在240 bp 缺失,缺失的原因可能是:同源修复后,Cas9 核酸酶对靶向区域的二次切割,鉴定含目标突变的克隆点为10% (5/50),点突变编辑效率偏低。YANG 等[25]和WANG 等[26]对成纤维细胞中点突变的编辑效率分别为11.4% (21/184) 和5.6% (5/90),这可能是受细胞类型、周期、同源重组模板质粒递送效率等因素的影响,导致细胞内同源重组修复途径发生的概率较低,造成点突变基因编辑效率也偏低,后续试验将对这些影响因素进行进一步的研究。
通过在转染过程中添加DNA ligase IV 抑制剂(SCR7)[27]、在同源修复模板上的PAM 结构引入同义突变改变ssODN 上的PAM 结构[28]、使用RNP 体系(Cas9 蛋白与体外转录的sgRNA) 代替质粒基因编辑工具[29-30]等方法可以有效提高基因编辑点突变的发生频率。在克隆点鉴定时,有5 个单克隆虽然含有目标点突变,但测序结果出现大量套峰和杂峰,表明该克隆点可能含有多种基因型。这是由于本研究使用药物筛选和有限稀释的方式进行单克隆培养和挑取,细胞间出现粘连所致。将药物筛选标记换成荧光基团,使用流式细胞仪进行单细胞分选,或许能改善粘连的状况,这将在后续的研究中进行验证。此外,针对p53 点效率偏低的问题,后续研究将改进同源修复突变模板,突变修复模板ssODN 中的PAM 结构,避免Cas9 核酸酶对靶向区域的二次切割;同时,在转染结束后向培养液中添加SCR7,抑制非同源性末端连接,提高同源重组概率,最终利用CRISPR/Cas9 技术,结合构建内源性p53V149F点突变基因修饰成纤维细胞系,通过核移植获得基因修饰的动物模型。
4 结论
本研究运用CRISPR/Cas9 基因编辑技术构建了有效的基因编辑质粒,并通过细胞水平的验证和T7EN1 酶切试验验证了该质粒的有效性;通过编辑后的细胞基因组测序结果设计了重组模板ssODN,经共转染基因编辑质粒和ssODN、药物筛选以及单克隆培养获得含有目标突变的细胞系,证明了利用CRISPR/Cas9 基因编辑技术靶向p53基因发生单碱基替换(G→T) 的定点精确编辑是切实可行的。
[ 参考文献 ]
[1]MA Y W, ZHANG L F, HUANG X X. Genome modificationby CRISPR/Cas9[J]. FEBS Journal, 2014, 281(23):5186. DOI: 10.1111/febs.13110.
[2]ZHAO Y C, DAI Z, LIANG Y, et al. Sequence-specificinhibition of microRNA via CRISPR/CRISPRi system[J].Scientific Reports, 2014, 4(1): 3943. DOI: 10.1038/srep03943.
[3]YANG L H, GÜELL M, NIU D, et al. Genome-wide inactivationof porcine endogenous retroviruses (PERVs)[J].Science, 2015, 350(6264): 1101. DOI: 10.1126/science.aad1191.
[4]ZHOU X Q, XIN J G, FAN N N, et al. Generation ofCRISPR/Cas9-mediated gene-targeted pigs via somaticcell nuclear transfer[J]. Cellular and Molecular Life Sciences,2015, 72(6): 1175. DOI: 10.1007/s00018-014-1744-7.
[5]HONDA A, HIROSE M, SANKAI T, et al. Single-stepgeneration of rabbits carrying a targeted allele of the tyrosinasegene using CRISPR/Cas9[J]. Experimental Animals,2015, 64(1): 31. DOI: 10.1538/expanim.14-0034.
[6]ZHAN T Z, RINDTORFF N, BETGE J, et al. CRISPR/Cas9 for cancer research and therapy[J]. Seminarsin Cancer Biology, 2019, 55: 106. DOI: 10.1016/j.semcancer.2018.04.001.
[7]ROY P S, SAIKIA B J. Cancer and cure: a critical analysis[J]. Indian Journal of Cancer, 2016, 53: 441. DOI:10.4103/0019-509x.200658.
[8]YIN W, WANG J L, JIANG L L, et al. Cancer and stemcells[J]. Experimental Biology and Medicine, 2021, 246:1791. DOI: 10.1177/15353702211005390.
[9]KEOHAVONG P, LAN Q, GAO W M, et al. Detectionof p53 and K-ras mutations in sputum of individuals exposedto smoky coal emissions in Xuanwei County, China[J]. Carcinogenesis, 2005, 26(2): 303. DOI: 10.1093/carcin/bgh328.
[10]LEROY B, ANDERSON M, SOUSSI T. TP53 mutationsin human cancer: database reassessment and prospectsfor the next decade[J]. Human Mutation, 2014,35(6): 672. DOI: 10.1002/humu.22552.
[11]KAUR R P, VASUDEVA K, KUMAR R, et al. Role ofp53 gene in breast cancer: focus on mutation spectrumand therapeutic strategies[J]. Current Pharmaceutical Design,2018, 24(30): 3566. DOI: 10.2174/1381612824666180926095709.
[12]YAEGER R, CHATILA W K, LIPSYC M D, et al. Clinicalsequencing defines the genomic landscape of metastaticcolorectal cancer[J]. Cancer Cell, 2018, 33(1): 125.DOI: 10.1016/j.ccell.2017.12.004.
[13]SONG Y M, LI L, OU Y W, et al. Identification of genomicalterations in oesophageal squamous cell cancer[J].Nature, 2014, 509(7498): 91. DOI: 10.1038/nature13176.
[14]LIEBL M C, HOFMANN T G. The role of p53 signalingin colorectal cancer[J]. Cancers, 2021, 13(9): 2125.DOI: 10.3390/cancers13092125.
[15]GADEPALLI V S, DEB S P, DEB S, et al. Lung cancerstem cells, p53 mutations and MDM2[M]//DEB S P, DEBS. Mutant p53 and MDM2 in Cancer. Dordrecht: Springer,2014.
[16]LI D, MARCHENKO N D, MOLL U M. SAHA showspreferential cytotoxicity in mutant p53 cancer cells bydestabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis[J]. Cell Death and Differentiation,2011, 18(12): 1904. DOI: 10.1038/cdd.2011.71.
[17]SUH Y A, POST S M, ELIZONDO-FRAIRE A C, et al.Multiple stress signals activate mutant p53 in vivo[J]. CancerResearch, 2011, 71(23): 7168. DOI: 10.1158/0008-5472.Can-11-0459.
[18]PETITJEAN A, MATHE E, KATO S, et al. Impact ofmutant p53 functional properties on TP53 mutation patternsand tumor phenotype: lessons from recent developmentsin the IARC TP53 database[J]. Human Mutation,2007, 28: 622. DOI: 10.1002/humu.20495.
[19]DUFFY M J, SYNNOTT N C, O’GRADY S, et al. Targetingp53 for the treatment of cancer[J]. Seminars inCancer Biology, 2022, 79(8): 58. DOI: 10.1016/j.semcancer.2020.07.005.
[20]OLIVIER M, HOLLSTEIN M, HAINAUT P. TP53mutations in human cancers: origins, consequences, andclinical use[J]. Cold Spring Harbor Perspectives in Biology,2010, 2(1): a001008. DOI: 10.1101/cshperspect.a001008.
[21]SAALFRANK A, JANSSEN K P, RAVON M, et al. Aporcine model of osteosarcoma[J]. Oncogenesis, 2016,5(3): e210. DOI: 10.1038/oncsis.2016.19.
[22]PRINCIPE D R, OVERGAARD N H, PARK A J, et al.KRASG12D and TP53R167H cooperate to induce pancreaticductal adenocarcinoma in Sus scrofa pigs[J]. Scientific Reports, 2018, 8(1): 12548. DOI: 10.1038/s41598-018-30916-6.
[23]SIEREN J C, MEYERHOLZ D K, WANG X J, et al.Development and translational imaging of a TP53 porcinetumorigenesis model[J]. Journal of Clinical Investigation,2014, 124(9): 4052. DOI: 10.1172/jci75447.
[24]DENISSENKO M F, CHEN J X, TANG M S, et al.Cytosine methylation determines hot spots of DNAdamage in the human P53 gene[J]. Proceedings of theNational Academy of Sciences of the United States ofAmerica, 1997, 94(8): 3893. DOI: 10.1073/pnas.94.8.3893.
[25]YANG Y, WANG K P, WU H, et al. Genetically humanizedpigs exclusively expressing human insulin are generatedthrough custom endonuclease-mediated seamlessengineering[J]. Journal of Molecular Cell Biology, 2016,8(2): 174. DOI: 10.1093/jmcb/mjw008.
[26]WANG K K, TANG X C, LIU Y, et al. Efficient generationof orthologous point mutations in pigs via CRISPRassistedssODN-mediated homology-directed repair[J].Molecular Therapy Nucleic Acids, 2016, 5(11): e396.DOI: 10.1038/mtna.2016.101.
[27]RAY U, VARTAK S V, RAGHAVAN S C. NHEJ inhibitorSCR7 and its different forms: promising CRISPRtools for genome engineering[J]. Gene, 2020, 763(15):144997. DOI: 10.1016/j.gene.2020.144997.
[28]张楷, 刘蔚, 刘小凤, 等. 利用CRISPR/Cas9系统构建人HPRT1基因定点突变细胞株[J]. 遗传, 2019, 41(10): 939.DOI: 10.16288/j.yczz.19-108.
[29]FU Y W, DAI X Y, WANG W T, et al. Dynamics andcompetition of CRISPR-Cas9 ribonucleoproteins and AAVdonor-mediated NHEJ, MMEJ and HDR editing[J].Nucleic Acids Research, 2021, 49(2): 969. DOI: 10.1093/nar/gkaa1251.
[30]许永汉, 齐泽宇, 李文静, 等. 精准序列替换基因组编辑技术研究进展[J]. 云南农业大学学报(自然科学), 2024,39(2): 162. DOI: 10.12101/j.issn.1004-390X(n).202302027.
责任编辑:何承刚
基金项目:云南省重大科技专项计划项目(202102AA100054)。
猜你喜欢
疯狂英语·新读写(2021年10期)2021-12-07 02:41:30
家庭医学(下半月)(2020年1期)2020-05-11 02:05:32
奥秘(2019年8期)2019-08-28 01:47:05
海峡姐妹(2018年7期)2018-07-27 02:30:36
特别健康(2018年4期)2018-07-03 00:38:08
特别健康(2018年2期)2018-06-29 06:13:42
商周刊(2017年7期)2017-08-22 03:36:21
今日健康(2016年11期)2017-06-09 03:02:11
科技视界(2017年2期)2017-04-18 18:52:33
小猕猴智力画刊(2016年6期)2016-05-14 09:21:40
