
水稻—马铃薯轮作对盆栽土壤细菌群落结构的影响
2024-08-04 00:00:00周远平王琼胡明举郭华春
云南农业大学学报(自然科学) 2024年3期
关键词:水稻




摘要: 【目的】研究耕作系统对土壤微生物群落的调节能力,为合理布局耕作系统奠定基础。【方法】以马铃薯连作为对照种植系统,在4 种条件下(土壤灭菌加施肥、土壤灭菌不施肥、土壤不灭菌加施肥、土壤不灭菌不施肥) 设计盆栽试验。通过PCR 技术扩增并测序水稻—马铃薯轮作(以下简称“稻薯轮作”) 和马铃薯连作系统中每个种植季节作物根区土壤样本的细菌16S rRNA 基因的V3~V4 区域,并比较细菌群落的物种组成、多样性、功能组成和生态网络。【结果】稻薯轮作可显著影响土壤细菌群落结构。在未灭菌条件下,轮作施肥和未施肥土壤的细菌群落丰富度指数分别显著提高15.62% 和9.05%,香农多样性指数分别显著提高2.45% 和1.71%;而连作土壤细菌群落的物种多样性和丰富度无显著变化。在土壤灭菌后,稻薯轮作土壤细菌群落物种的丰富度指数和香农多样性指数可提高到灭菌前水平;而连作土壤细菌群落物种的香农多样性指数显著降低,丰富度仅在施肥时恢复到土壤灭菌前水平。稻薯轮作和马铃薯连作土壤细菌群落功能组成差异分析表明:稻薯轮作可显著提高13 类土壤功能菌群的丰度;而连作仅能提高4 类土壤功能菌群的丰度,其中包括几丁质分解菌和植物病原菌。与连作相比,轮作土壤细菌群落生态网络的介数中心性、边数、图密度、节点数、平均度、图径、模块性和平均路径长度分别提高了10.00 倍、2.50 倍、2.40 倍、1.10 倍、0.64 倍、0.63 倍、0.55 倍和0.40 倍;而细菌病原菌属群体生态网络的介数中心性、图密度、边数、平均度、节点数和度中心性分别降低了71.4%、67.7%、50.9%、36.1%、23.2% 和11.1%。此外,冬季马铃薯种植土壤的病原菌属群体物种的生态网络拓扑特征参数也较夏季耕作土壤显著降低。【结论】稻薯轮作可显著提高土壤细菌群落的物种和功能多样性及调控土壤细菌群落物种的相互作用,对土壤细菌群落具有显著的调节作用。
关键词: 水稻—马铃薯轮作;马铃薯连作;细菌物种多样性;功能组成;生态网络结构
中图分类号: S344.17 文献标志码: A 文章编号: 1004–390X (2024) 03−0001−16
中国冬季约有1.6×107 hm2 冬闲稻田[1],合理利用冬闲稻田可为粮食安全作出巨大贡献。由于马铃薯(Solanum tuberosum L.) 喜好冷凉生长环境,在冬闲稻田大力推广水稻—马铃薯轮作(以下简称“稻薯轮作”) 种植模式具有良好的发展前景。然而,田间作物生长常受到土壤细菌群落的影响,评估和探明稻薯轮作模式对土壤细菌群落的调节效应,可为土壤微生物群落的管理提供理论依据。
土壤细菌群落是抑制土传病菌[2-5]、促进土壤有机物和无机物生物地球化学循环[6-7]等生态系统服务功能的资源库。连作生产引起土传病害暴发,可造成严重的经济损失[8]。土壤微生物群落中,病原菌属中的有益菌群常常发挥病菌抑制的关键作用[5, 9-11],且病原菌属内的有益菌可能与致病菌形成互作关系。因此,利用耕作系统调节土壤菌群结构、防控土传病害是重要的农业措施[12]。前人对间作[13-15]和轮作系统[16-18]的研究与实践,减轻了土传病菌造成的损失[19-20],表明较好的耕作系统中微生物群落物种多样性较高,且病原菌的16S 基因拷贝数丰度较低。鉴于目前还无准确鉴定土壤环境中作物致病菌的高通量方法,本研究结合细菌群落物种多样性、功能多样性、共生网络稳定性等微生态指标,深入分析了稻薯轮作较马铃薯连作的土壤微生态调节优势,为定性稻薯轮作系统的生态效应提供理论依据。
利用扩增子测序数据构建微生物生态网络,可分析微生物群落潜在的种间互作模式[7, 21-23]。宿主根系微生物菌群由有益菌群和有害菌群共同组成[24],且用扩增子技术可实现在菌属水平上检测前人已报道的病原菌属(包含致病菌和有益菌),再加上扩增子技术以ASVs (扩增序列变体) 代替OTUs 的方法提高了对微生物群落物种的分辨率[25],可将耕作系统对土壤微生物群落的影响聚焦到病原菌属群体,进而利用网络分析表征病原菌属微生物群体内的互作模式对耕作系统的响应,可明确病原菌属中微生物群体的种间互作模式在耕作系统间的差异特征,为在群体水平上分析不同耕作系统的微生物群落结构差异提供新思路。然而,目前尚未有以此视角分析耕作系统间物种生态网络差异的研究。
前人研究了稻薯轮作对土壤疮痂病菌的抑制效果[26],该研究利用平板培养菌落计数法比较了基于稻薯轮作的不同耕作系统中土壤疮痂病菌数量的差异,认为在休闲期种植绿肥作物或采用休耕措施可抑制疮痂病菌,但种植水稻对土壤疮痂病菌群体的效应还有待深入研究,且平板培养计数法对土壤微生物群体的调查不够全面[27]。此外,施肥是农业生产中的重要措施,干扰土壤原有菌群可影响作物生长,因此,本研究设置施肥、土壤灭菌等处理,深入分析灭菌、施肥、种植季节、耕作模式等因素对土壤微生物群落结构的影响,揭示稻薯轮作模式对土壤细菌群落的调控机理。
1 材料与方法
1.1 试验材料
供试材料为从稻薯轮作处理和马铃薯连作处理中采集的作物根区土壤,具体处理见1.1.1 节。
1.1.1 试验设计
本研究模拟稻薯轮作系统[1], 在云南农业大学薯类作物研究所试验地(25°8′15″N, 102°45′5″E)设计稻薯轮作和马铃薯连作模式的对比盆栽试验。在温室内采集0~20 cm 土层土壤,混匀后依次分装土壤25 kg 至80 个盆(不漏水, 直径30 cm×高45 cm) 内作为后续作物种植的土壤基质;采集3 份混匀土壤样品作为细菌群落结构分析的对照土壤样品(CK,3 个重复)。将80 盆土壤随机平均分为2 份,其中40 盆土壤单独装入透明塑料袋中,用高压灭菌锅121 ℃ 灭菌45 min 后重新装回盆内。按照试验设计(耕作模式×土壤灭菌×土壤施肥×种植季节) 将80 盆土壤摆放于大棚内(图1),其中,施肥处理的施肥量为每盆有机肥(综合肥力≥5%) 500 g,并按尿素(含纯氮46.4%)2.320 g、过磷酸钙(P2O5 含量16%) 5.874 g、马铃薯专用肥(N-P2O5-K2O 含量为10%-8%-18%)7.750 g 的施肥水平施用化学肥料。施肥水平与2011—2015 年间水稻和马铃薯的田间施肥量相当。选择由云南农业大学水稻作物研究所育成的水稻品种滇杂36 和马铃薯品种青薯9 号为供试作物。以青薯9 号连作2 茬(包括夏作和冬作)、水稻滇杂36—青薯9 号轮作1 轮(2 茬,包括夏作和冬作) 进行对比试验。马铃薯和水稻夏作时间为2016 年4 月18 日,马铃薯冬作时间为2016年11 月20 日;其中,水稻育秧时间为2016 年4 月18 日—2016 年6 月10 日,插秧时间为2016年6 月10 日。连作土壤盆栽中,每盆土壤播种1 块健康且大小一致的块茎,轮作土壤中移栽1 株健康且大小一致的水稻三叶稻苗。马铃薯和水稻夏作作物分别于2016 年8 月7 日和2016 年10 月13 日收获,冬作马铃薯于2017 年4 月3 日收获。作物生长期间,及时拔除杂草并根据作物需水量浇水。
1.1.2 土壤样品采集及保存
于每季作物(夏季水稻或马铃薯及冬季马铃薯) 收获时,戴上手套和口罩,用不锈钢铲将作物根系取出,采用抖落法将非根区土壤抖回盆内,用灭菌毛刷(每个处理单独使用毛刷) 收集紧密结合在作物根系表面的土壤于灭菌保鲜膜上,再用灭菌的不锈钢勺转移至50 mL 无菌离心管中,用于提取土壤DNA。共采集了17 个处理的51份土壤样品, 分别标记为CK、 CCAFs、 CCAnFs、CCunAFs、CCunAnFs、RCAFs、RCAnFs、RCun-AFs、RCunAnFs、CCAFw、CCAnFw、CCunAFw、CCunAnFw、RCAFw、RCAnFw、RCAnFw 和RCunAnFw(CC 表示连作,RC 表示轮作,A 表示灭菌,unA 表示未灭菌,F 表示施肥,nF 表示未施肥,s 表示夏作,w 表示冬作),置于冰上送至实验室存储于−80 ℃ 冰箱备用。
1.2 土壤细菌群落16S rRNA 基因V3~V4 区测序
1.2.1 DNA 提取和PCR 扩增
遵循E.Z.N.A®土壤DNA 试剂盒(Omega Biotek,Norcross,GA,USA) 的操作手册提取土壤样品总DNA。先用1% 琼脂糖凝胶检测DNA 质量,然后用NanoDrop 2000 UV-vis 分光光度计(Thermo Scientific,Wilmington,USA) 检测样品DNA 浓度和纯度。以提取的DNA 为模板,采用PCR 技术以引物对338F (5′-ACTCCTACGGGAGGCAGCAG-3′) 和806R (5′-GGACTACHVGGGTWTCTAAT-3′) 在ABI GeneAmp®9700 PCR 仪(ABI,CA,USA) 上扩增细菌16S rRNA 基因V3~V4 高变区。PCR 反应体系为:5×TransStart Fast-Pfu 缓冲液4.0 μL, 2.5 mmol/L dNTPs 2.0 μL,5 μmol/L 正、反向引物各0.8 μL,TransStart FastPfuDNA 聚合酶0.4 μL,模板DNA 10 ng,加ddH2O至20.0 μL。PCR 扩增程序为: 95 ℃ 预变性3 min,27 个循环的95 ℃ 变性30 s,55 ℃ 退火30 s,72 ℃ 延伸45 s,随后72 ℃ 延伸10 min,并于4 ℃ 保存。每个样品PCR 扩增3 次;PCR 终产物按照AxyPrep DNA 胶提取试剂盒(AxygenBiosciences,Union City,CA,USA) 操作指南,先用2% 琼脂糖凝胶提取纯化,再用QuantusTMFluorometer (Promega,USA) 定量。
1.2.2 Illumina Miseq 测序
扩增子(PCR 产物) 等摩尔混样后,利用IlluminaMiseq PE300 平台(Illumina,San Diego,USA)双端测序。原始测序序列质控后,用FLASH 软件按如下规则拼接:(1) 过滤reads 尾部质量值小于20 的碱基,设置50 bp 的窗口,平均质量值低于20,则截去后端碱基,舍弃质控后小于50 bp的reads 以及含N 碱基的reads;(2) 根据序列间的重叠关系,将序列拼接成1 条,最小重叠为10 bp;(3) 允许拼接序列重叠区的最大错配比率为0.2,剔除不满足条件的序列;(4) 根据序列首尾两端的条码和引物区分样品,并调整序列方向,条码允许的错配数为0,最大引物错配数为2。从上海美吉生物科技有限公司获得测序数据后,将样本扩增子纯净序列上传至GSA 数据库(Genome Sequence Archive-CNCB-NGDC) 中(登录号为:CRA006528)。
1.2.3 序列数据处理
进一步舍弃纯净序列中序列数少于10 条的序列,以99% 为阈值,利用USEARCH v10.0 软件[28]中的unoise3 算法对序列变体进行聚类。利用VEARCH v2.15.0 软件[29]中Uchime 算法,基于参考数据库的方法检测并移除嵌合体序列。基于SILVA v123 数据库,每种序列变体的代表性序列用USEARCH v10.0 软件的SINTAX 模块、以0.6 为阈值进行分类注释。利用pipeline[30]中的R 代码将样本序列等量重抽样至与序列数最少的样本一致(12 634 条序列),供后续细菌群落α 和β 多样性分析。
1.3 细菌群落物种生态网络构建及分析
1.3.1 细菌群落生态网络构建及其拓扑特征计算
利用R 软件包SpiecEasi v1.1.2[23],以获得的扩增序列变体构建细菌稀疏性生态网络。移除特征表中的对照样本数据,将剩余样本数据分别依据耕作系统(连作和轮作)、灭菌(灭菌和未灭菌)、施肥(施肥和未施肥)、季节(夏作和冬作)等处理进行拆分,得到8 个子处理特征表;对每个子处理特征表进行总丰度排序,先选出丰度排名前400 的ASV,随后基于文献报道的病原菌属[10]再选出病原菌属中丰度排名前400 的ASV,共得到16 个子处理特征表,用于构建细菌群落生态网络。利用R 包Phyloseq v1.36.0 合并16 个子处理特征表、样本数据采集信息表及与子处理特征表对应的物种分类注释表,以满足SpiecEasi 包网络构建的格式;利用SpiecEasi 包中的glasso模块和StARS 参数,构建不同处理细菌群落的生态网络,StARS 变化阈值设定为0.01。利用R 包igraph v1.2.6 计算各生态网络拓扑特征。
1.3.2 模块检测及节点角色鉴定
分析不同子处理的细菌群落生态网络的模块和节点角色。1 个模块由在组内有较高连通度而在组外几乎无连通度的1 组节点组成,其中快速贪婪模块优化算法(fast greedy modularity optimization)[22]被用于模块检测,依据文献[31-32] 报道,以模块系数gt;0.4 为模块判定阈值。其中,每个节点的角色依据模块内连通度(Zi) 和模块间连通度(Pi) 分类,可分为4 类:参与节点,即与模块中节点彼此相连却很少与模块外节点相连的节点,其Zilt;2.5 且Pilt;0.62;模块节点,即与模块内节点高度连接的节点,其Zigt;2.5 且Pilt;0.62;连接节点,即连接不同模块的节点,其Zilt;2.5 且Pigt;0.62;网络节点,即在整个网络中具有高连通度的节点,其Zigt;2.5 且Pigt;0.62。
1.4 土壤细菌群落物种功能组成注释
将细菌群落物种组成分析流程生成的特征表进行重抽样,结合物种分类注释表,依据FAPROTAXv1.2.5 数据库[33]对物种功能组成进行分类,用于比较不同处理间群落物种功能的差异。
1.5 数据处理
除细菌群落物种功能组成差异分析外,均采用R v4.0.2 软件分析不同处理间菌群结构差异,显著性水平默认为α=0.05,具体步骤为:先利用amplicon 包的单因素方差分析模块分析不同处理土壤细菌群落α 物种多样性(多样性指数和丰富度指数) 差异,再用Tukey-HSD 算法进行post-hoc检验;利用vegan v2.5.6 包的adonis2 函数对细菌群落结构进行多元方差分析,以量化不同处理因素对细菌群落物种组成的独立效应;利用randomForestv4.7.1.1 包分析细菌群落物种功能组成在不同处理间的重要性,选取最重要的前20 类功能组成,用STAMP v2.1.3 软件中的Welch’st检验进一步分析不同处理对土壤细菌群落物种功能组成的影响,并可视化分析结果。
2 结果与分析
2.1 土壤样品中细菌群落的优势分类群
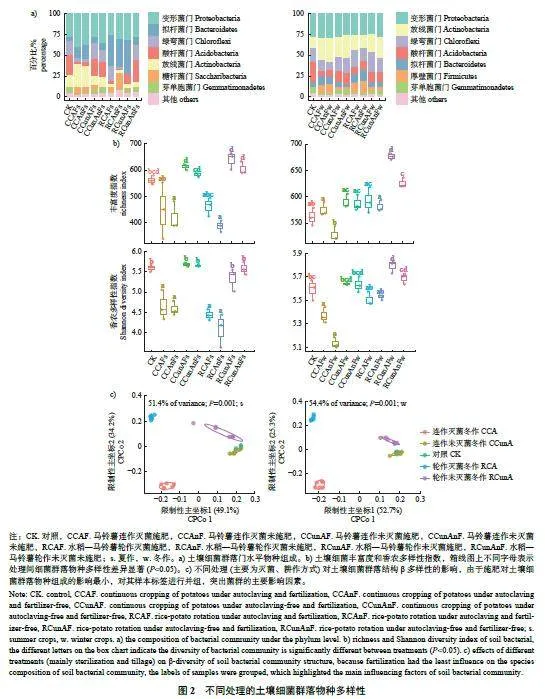
共检测到12 727 个扩增序列变体,隶属于26 门51 纲84 目159 科218 属。门水平上,变形菌门(Proteobacteria,31.9%)、放线菌门(Actinobacteria,15.3%)、绿弯菌门(Chloroflexi,14.0%)、酸杆菌门(Acidobacteria,12.7%)、拟杆菌门(Bacteroidetes,11.2%) 和厚壁菌门(Firmicutes,5.3%)的平均丰度最高;属水平上,10 个高丰度菌属分别为鞘氨醇单胞菌属(Sphingomonas,2.4%)、芽孢杆菌属(Bacillus,1.6%)、溶杆菌属(Lysobacter,1.6%)、Blastocatella (1.5%)、诺卡氏菌属(Nocardia,1.5%)、Flavisolibacter (1.5%)、 Roseiflexus(1.4%)、布氏菌属(Bryobacter,1.4%)、链霉菌属(Streptomyces,1.3%) 和芽单胞菌属(Gemmatimonas,1.0%)。其中,种植马铃薯可提高放线菌门细菌的物种丰度;种植水稻可急剧提高拟杆菌门细菌的物种丰度、急剧降低放线菌门的物种丰度(图2a)。
2.2 细菌α 和β 多样性
经马铃薯或水稻夏作后,与种植前对照土壤(CK) 相比,灭菌土壤细菌群落物种多样性呈降低趋势;在未灭菌土壤中,细菌群落物种多样性较CK 无显著变化,且连作土壤和轮作土壤之间细菌群落物种多样性也无显著差异(图2b)。
马铃薯冬作后,在稻薯轮作土壤中,灭菌土壤细菌群落的物种多样性恢复到与CK 相当的水平;而在马铃薯连作土壤中,仅细菌群落物种丰富度在施肥条件下恢复到CK 水平,而未施肥条件下的细菌群落物种丰富度仍低于CK;无论施肥与否,细菌群落物种香农多样性指数均显著低于CK。在未灭菌土壤中,轮作施肥土壤的细菌群落物种多样性较CK 显著提高,且轮作未施肥土壤细菌群落的物种丰富度也显著高于CK;轮作施肥土壤细菌的群落丰富度显著高于马铃薯连作施肥土壤(图2b)。
由表1 和图2c 可知:土壤灭菌、耕作模式和施肥均能显著影响土壤细菌群落物种组成(Plt;0.05),且轮作和灭菌是两大主要影响因素。
2.3 细菌群落生态网络对不同处理的响应
2.3.1 细菌群落生态网络拓扑结构特征对不同处理的响应
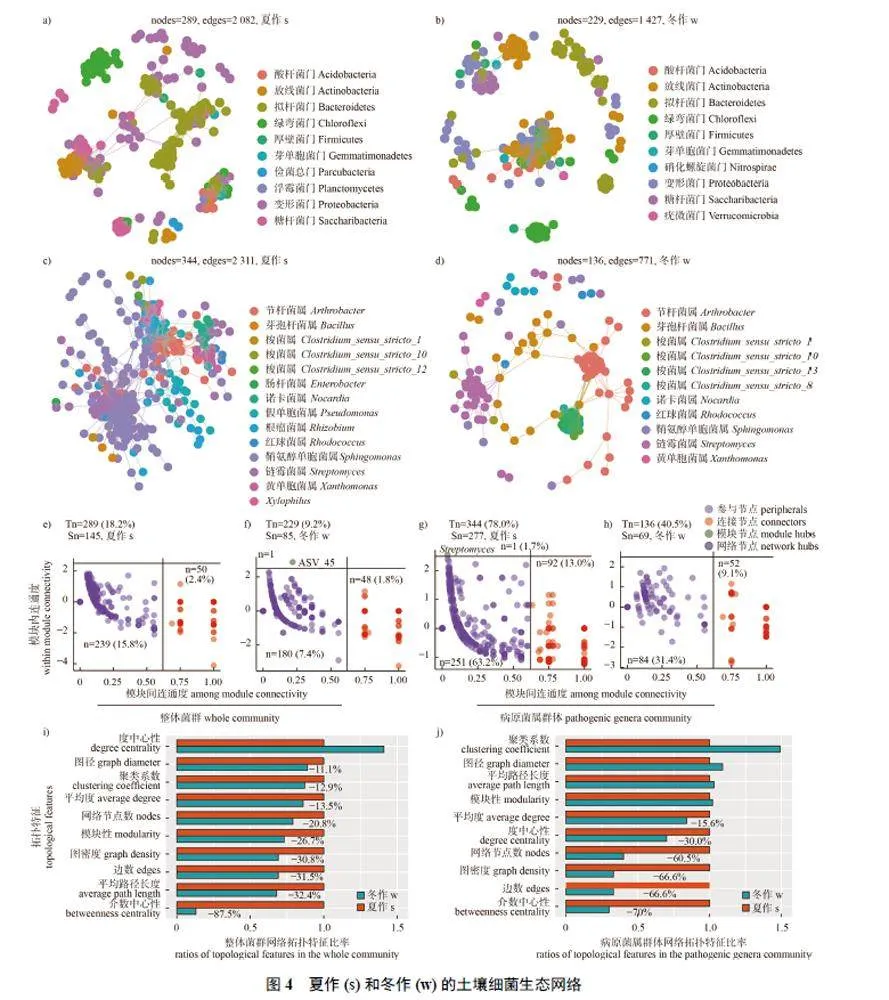
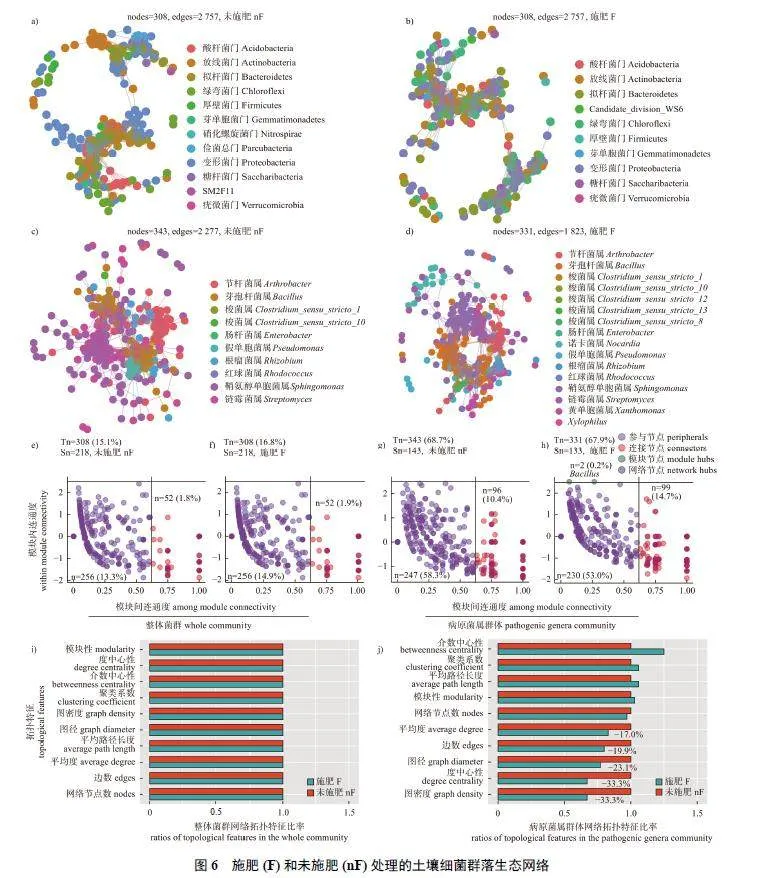
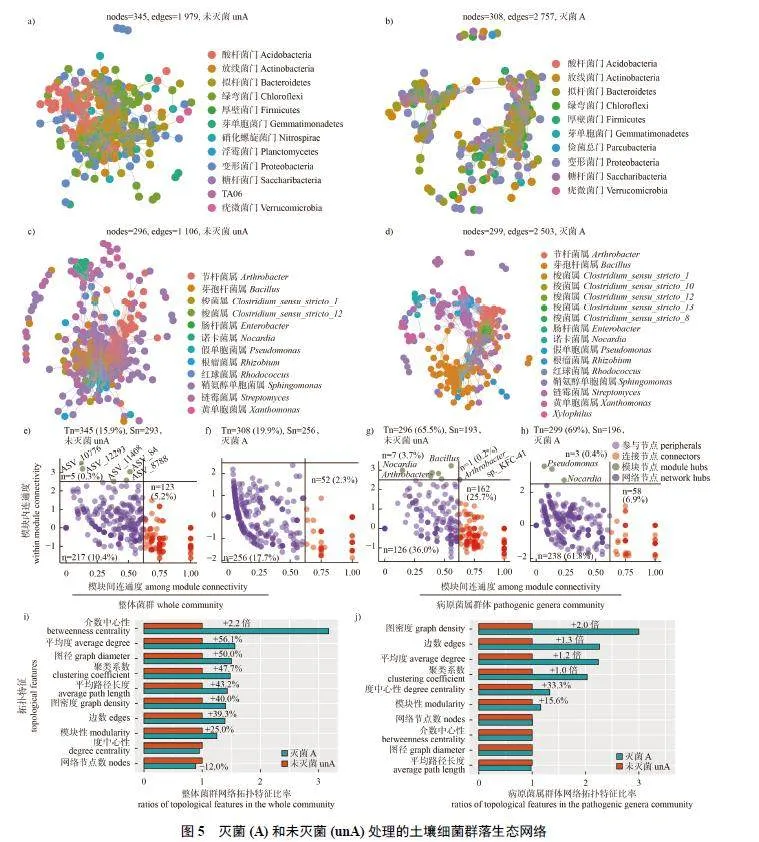
以土壤细菌群落中高丰度优势菌群表征生态网络结构对不同处理的响应,结果显示:连作土壤菌群网络复杂度远低于轮作土壤(图3a 和3b);夏作土壤菌群网络结构复杂度略高于冬作土壤(图4a 和4b);灭菌土壤网络节点数略低于未灭菌土壤,但其边数远高于未灭菌土壤(图5a 和5b);施肥土壤和未施肥土壤菌群网络结构复杂度相似(图6a 和6b)。以病原菌属群体中优势菌群表征生态网络结构对不同处理的响应,结果表明:连作土壤形成了复杂度远高于轮作土壤的细菌菌群网络结构(图3c 和3d),这与整体菌群构建的菌群网络结果相反;冬作土壤菌群网络结构复杂度远低于夏作土壤(图4c 和4d);灭菌土壤形成了较未灭菌土壤边数更多的复杂网络结构(图5c 和5d);而未施肥土壤菌群网络结构复杂度略高于施肥土壤(图6c 和6d)。
2.3.2 细菌群落生态网络节点角色对不同处理的响应
以土壤细菌群体中高丰度优势菌群表征生态网络节点角色对不同处理的响应,结果显示:马铃薯连作土壤网络的参与节点数和相对丰度均明显低于稻薯轮作土壤,且具有1 个模块节点(图3e和3f);夏作土壤网络的参与节点数和相对丰度略高于冬作土壤(图4e 和4f);灭菌土壤的网络模块节点数、连接节点数和相对丰度明显低于未灭菌土壤,而参与节点数及丰度明显高于未灭菌处理(图5e 和5f);施肥和未施肥土壤网络间的节点角色相似(图6e 和6f)。
以病原菌属中高丰度菌群生态网络表征细菌生态网络节点角色对不同处理的响应,结果显示:马铃薯连作土壤的模块节点和参与节点数量更多、丰度更高,而连接节点数量更少、丰度更低,其丰度排名前400 的ASV 群体中网络节点和边的数量及丰度远高于稻薯轮作土壤,其网络模块节点分别属于芽孢杆菌属和节杆菌属(Arthrobacter)细菌,而轮作土壤网络的模块节点属于梭菌属(Clostridium sensu stricto) 细菌(图3g 和3h),说明不同种植模式下土壤细菌群落中的功能菌群存在显著差异。夏作土壤网络的模块节点、连接节点和参与节点的数量及丰度均远高于冬作土壤,但其唯一的模块节点属于链霉菌属细菌(图4g 和4h),是马铃薯疮痂病的致病菌属。未灭菌土壤较灭菌土壤网络的组成节点分为4 类,不仅含有1 个网络节点,且其模块节点和连接节点的数量更多且丰度更高,但参与节点的数量较少且丰度较低,其网络节点和模块节点分别属于节杆菌属、诺卡氏菌属和芽孢杆菌属细菌;而灭菌土壤的模块节点分别属于诺卡氏菌属和假单胞菌属(Pseudomonas) 细菌(图5g 和5h)。施肥和未施肥土壤菌群网络的节点角色相似,在施肥土壤中有2 个模块节点,均属于芽孢杆菌属细菌(图6g和6h)。
此外,菌群生态网络拓扑结构特征(图3i 和3j、图4i 和4j、图5i 和5j、图6i 和6j) 与菌群生态网络结构表现一致,在各处理土壤的病原菌属物种群体生态网络中,属内或属间节点互作关系频繁,且在处理间表现出明显的差异特征。
2.4 细菌功能组成对不同处理的响应
由图7 可知:土壤灭菌处理显著提高了土壤中参与需氧化能异养(C 化合物代谢)、发酵(C 化合物代谢)、甲基化(C 化合物代谢)、甲醇氧化(C 化合物代谢)、尿素分解(N 化合物代谢)、硝酸盐氨化(N 化合物代谢)、砷酸盐呼吸(As 化合物代谢)、砷酸盐异化还原(As 化合物代谢) 等生化过程的细菌群落物种相对丰度;而未灭菌处理显著提高了土壤中参与硝化(N 化合物代谢)、好氧氨氧化(N 化合物代谢)、好氧亚硝酸盐氧化(N 化合物代谢)、捕食和体外寄生(C 化合物代谢)、硫呼吸(S 化合物代谢)、光能异养(C 化合物代谢) 等生化过程的细菌群落物种相对丰度(图7a)。施肥显著提高了土壤中参与硫化合物的暗氧化(S 化合物代谢)、硫酸盐呼吸(S 化合物代谢)、暗硫氧化(S 化合物代谢)、碳氢化合物降解(C 化合物代谢)、脂肪族非甲烷烃类降解(C 化合物代谢)、芳香烃降解(C 化合物代谢)、暗铁氧化(Fe 化合物代谢) 等生化过程的细菌群落物种相对丰度;而未施肥土壤显著提高了参与捕食和体外寄生(C 化合物代谢)、固氮(N 化合物代谢)、甲烷合成(C 化合物代谢) 等生化过程的细菌群落物种相对丰度(图7b)。马铃薯连作显著提高了土壤中参与需氧化能异养(C 化合物代谢)、化能异养(C 化合物代谢)、几丁质溶解(C 化合物代谢) 等生化过程以及植物病原菌(C 化合物代谢) 等细菌群落物种相对丰度;而稻薯轮作显著提高了土壤中参与暗硫化合物氧化(S 化合物代谢)、硫化合物呼吸(S 化合物代谢)、暗硫氧化(S 化合物代谢)、发酵(C 化合物代谢)、甲基化(C 化合物代谢)、甲烷合成(C 化合物代谢)、富马酸盐呼吸(C 化合物代谢)、铁呼吸(Fe 化合物代谢)、暗铁呼吸(Fe 化合物代谢)、胞内寄生(C化合物代谢)、亚硝酸盐呼吸(N 化合物代谢)、亚硝酸盐反硝化(N 化合物代谢)、氢暗氧化(H 化合物代谢) 等生化过程的细菌群落物种相对丰度(图7c)。夏作土壤中显著提高了土壤中与动物寄生或共生(C 化合物代谢) 的细菌群落物种相对丰度;而冬作土壤中显著提高了参与芳香族化合物降解(C 化合物代谢)、木质素分解(C 化合物代谢)、硫酸盐呼吸(S 化合物代谢)、硝酸盐氨化(N 化合物代谢)、亚硝酸盐氨化(N 化合物代谢)、锰氧化(Mn 化合物代谢)、砷酸盐呼吸(As 化合物代谢)、砷酸盐异化还原(As 化合物代谢) 等生化过程的细菌群落物种相对丰度(图7d)。
3 讨论
当前是探寻提高粮食产量和促进农业可持续生产的农业措施以应对2050 年粮食挑战的关键节点[34-35]。本研究的主要目的是探明与马铃薯连作相比,稻薯轮作能否调控土壤细菌群落中的病原菌群体,并解析其调控机制。由于马铃薯连作经常造成土传病害暴发[19, 36],而稻薯轮作具有巨大的增产潜力[1],因此,本研究以马铃薯连作模式为对照设计了温室盆栽试验,以分析稻薯轮作模式对土壤土传病原菌属菌群的影响;利用已有文献报道的植物病原菌属[10]中的高丰度物种[37]构建细菌群落物种生态网络,以表征病原菌群的种间互作关系对耕作模式、种植季节、灭菌和施肥的响应,结合不同处理中细菌群落的物种多样性、生态网络结构、潜在功能组成、作物细菌病原菌属菌群的生态网络结构等指标,综合分析稻薯轮作模式对土壤细菌菌群结构的影响。
3.1 稻薯轮作可显著提高土壤细菌群落的物种多样性
细菌群落物种多元方差分析表明: 耕作模式是影响土壤细菌群落结构的重要因素(R2≥0.139 1%),这与已有研究中“轮作可改变土壤细菌群落物种多样性”的观点[18]一致;此外,灭菌土壤中轮作较连作可提高土壤细菌群落物种多样性和丰富度,较高的香农多样性指数和丰富度指数表明稻薯轮作在维持或提升细菌群落物种多样性方面较马铃薯连作模式有优势,这一结果符合有关作物多样化种植系统的观点[38-39];本研究还表明:连作土壤灭菌条件下,其细菌群落丰富度需要施肥才能提高到与对照水平相当。以上结果表明:稻薯轮作及施肥措施在细菌群落物种多样性的恢复过程中发挥了关键作用。然而,由于稻薯轮作模式属于集约化耕作,故推荐在未来的研究和实践中对该耕作模式进行更为多样化的设计[26]并优化施肥方案,进一步结合微生物宏基因组学技术,明确多样化设计后稻薯轮作模式对土壤微生物群落物种多样性的影响,提高该轮作模式的生产效益。
3.2 细菌群落生态网络是不同农业生产措施的有效指标
微生物生态网络的主要效用是识别生态位中微生物群落物种的种间互作模式,并探索定向调控微生物群落的潜在有效手段或表征特定生态位中微生物群落物种的潜在种间互作关系[23, 40]。本研究利用SpeicEasi 包中的glasso 方法推断不同处理下土壤中细菌群落的生态网络结构特征,该方法在构建细菌群落生态网络时,相较于普遍采用基于相关性的方法,可在一定程度上消除网络中虚假的种间互作关系。此外,研究中仅采用不同处理下土壤样本中丰度排名前400 的ASV群体构建网络,其原因是网络构建耗时,而生态位中高丰度物种可反映整个群落物种的互作趋势[37, 40-42],且核心微生物群体对作物产量[41]和耕作系统的可持续性[40]具有重要作用,故在构建生态网络时赋予高丰度物种更高的权重。
本研究显示:稻薯轮作在高丰度菌群物种中形成了更为复杂和稳健的网络结构,而马铃薯连作则在细菌病原菌群体中形成了更复杂和更稳健的网络结构,即稻薯轮作对土壤细菌病原菌群有抑制作用[43],表明在病原菌属群体中构建菌群物种的生态网络结构表征病原菌群对农业措施的响应、对采纳合理的农业措施具有重要的参考价值。本研究表明:冬作土壤菌群网络受到抑制,这与前人研究结果[44]不尽一致,且本研究显示:冬作极大地消除了细菌病原菌群体的物种生态网络,这一结果为在冬闲稻田推广种植重要且适宜的细菌易感病作物时提供了重要的参考依据。此外,灭菌和未施肥土壤形成的生态网络分别较未灭菌和施肥土壤形成了更高的网络图密度和更多的边数,这可能与有益细菌群体对非生物逆境诱导的生物逆境(有益菌群对病菌入侵的防御)[45]和非生物逆境(未施肥) 响应有关。灭菌打破了土壤细菌群落生态网络中的模块节点和连接节点,却促进了参与节点的生成,表明参与节点在整个细菌群落中可能具有重要的生态功能;且灭菌土壤细菌群落的模块节点由未灭菌土壤中的节杆菌属和芽孢杆菌属细菌变成了假单胞菌属细菌,而已有研究表明假单胞菌属中的许多物种具有马铃薯疮痂病菌抑制效应[5],这种转变表明灭菌土壤中发生了菌群的抗病响应,这与土壤细菌群落的诱导抗性机制[45]较为吻合。本研究显示:稻薯轮作土壤较马铃薯连作土壤的病原菌群网络模块节点和参与节点数量少、丰度低,而连接节点数量多,表明马铃薯连作对土壤病原菌群的调控效应低于稻薯轮作[37]。值得注意的是,虽然施肥和未施肥土壤的细菌群落生态网络节点的角色较相似,但两者网络节点的物种组成极为不同,表明施肥也是有效调节土壤细菌群落结构的农业措施[18];但由于微生物群落中的物种间往往由于功能冗余的原因,导致施肥条件虽调节了土壤微生物的物种组成,但不一定影响微生物群体的网络菌群互作结构。与夏作相比,冬作可降低土壤细菌群落生态网络的参与节点数及相对丰度,明显降低病原菌群体生态网络中的模块节点、连接节点和参与节点的数量及相对丰度,且夏作土壤中唯一的模块节点为链霉菌属细菌,该属致病菌可严重为害马铃薯块茎,其中,马铃薯疮痂致病菌基因组中的致病岛基因可在属内物种间转移[46],使非致病菌可转变为致病菌。可见,冬作更有利于马铃薯生产。
本研究主要分析了细菌群落生态网络对不同处理的响应,但生态网络只能刻画土壤中微生物群落的相关关系,表征群落中特定菌群的互作模式以及不同处理中菌群互作模式的动态变化;而物种间的实际互作关系(竞争、合作、拮抗和共生) 需要进一步的共培养试验予以证明。由于群落中物种数量巨大,当前研究只能采取网络分析的方法探索微生物群落在土壤中的潜在互作关系,为后期验证重要的菌株互作模式提供参考[47]。利用细菌群落物种组成数据构建生态网络推断土壤原位微生物群落中潜在的种间互作关系、分析耕作模式对土壤微生物群落的调节机制,进而利用具有高重用性、基于ASV 的微生物群落表征技术,对大田农业管理措施具有重要的指导意义。
3.3 细菌代谢功能组成证实稻薯轮作的病原菌群调控机制
基于FAPROTAX v1.2.5 数据库[33]注释土壤细菌群落的物种潜在功能组成,结果表明:马铃薯连作显著促进了土壤中C 化合物代谢相关菌群的丰度,与前人对连作系统的研究结论[48-49]一致。尽管连作土壤C 代谢活动的几丁质溶解过程对植物病害具有抑制作用[50],但马铃薯连作较稻薯轮作仍显著提高了土壤中植物病原细菌的相对丰度。马铃薯连作土壤细菌群落的功能组成多样性显著低于稻薯轮作土壤,且马铃薯连作土壤的C 化合物代谢菌群丰度显著高于轮作土壤,进一步表明马铃薯连作极大地促进了其耕作土壤中C 素的分解,恶化了土壤环境,显著提高了植物细菌性病害群体的相对丰度,这可能是造成细菌病害暴发的重要原因。同时,夏作显著提高了土壤动物寄生或共生细菌的相对丰度,而冬作则显著提高了细菌群落的功能组成多样性,表明冬作可能为作物和微生物提供了生态功能良好的环境,限制了细菌的寄生或共生过程,促进了土壤中C 化合物和N 化合物的代谢,这与气温不影响土壤细菌功能多样性的结论[51]相反。
本研究通过细菌群落功能组成注释分析了不同处理对土壤细菌功能组成的影响,未来可结合本研究使用的病原菌群生态网络进行代谢物指标检测[52]和宏基因组测序[53],对稻薯轮作系统的土壤微生态效应进行全面且深入的研究,并基于本研究结论设计和优化更合理的稻薯轮作种植模式。
4 结论
与马铃薯连作相比,稻薯轮作可通过提升土壤细菌群落物种多样性和功能多样性降低土壤细菌群体中植物致病菌的相对丰度,提高土壤细菌群落物种微生态网络的复杂度和稳健性,进而抑制土传病原菌属细菌群体的物种微生态网络,降低致病菌群的物种丰度,发挥对土壤细菌群落中病原菌群体的调控作用。相较于夏作,冬作可打破土壤致病菌属中物种间的微生态网络结构,进而提升稻薯轮作系统对土传细菌病原菌群体的抑制作用,有利于防控冬闲稻田土壤中的土传细菌病害。
[ 参考文献 ]
[1]LU Y, KEAR P, LU X P, et al. The status and challengesof sustainable intensification of rice-potato systemsin southern China[J]. American Journal of Potato Research,2021, 98(5): 361. DOI: 10.1007/s12230-021-09848-x.
[2]GUO Y Q, LUO H, WANG L, et al. Multifunctionalityand microbial communities in agricultural soils regulatethe dynamics of a soil-borne pathogen[J]. Plant and Soil,2021, 461(1): 309. DOI: 10.1007/s11104-020-04826-4.
[3]CASTALDI S, CIMMINO A, MASI M, et al. Bacteriallipodepsipeptides and some of their derivatives and cyclicdipeptides as potential agents for biocontrol of patho-genic bacteria and fungi of agrarian plants[J]. Journal ofAgricultural and Food Chemistry, 2022, 70(15): 4591.DOI: 10.1021/acs.jafc.1c08139.
[4]ZHOU Y J, LI Q, PENG Z, et al. Biocontrol effect of Bacillussubtilis YPS-32 on potato common scab and itscomplete genome sequence analysis[J]. Journal of Agriculturaland Food Chemistry, 2022, 70(17): 5339. DOI:10.1021/acs.jafc.2c00274.
[5]PACHECO-MORENO A, STEFANATO F L, FORD J J,et al. Pan-genome analysis identifies intersecting rolesfor specialized metabolites in potato pathogen inhibition[J]. eLife, 2021, 10: e71900. DOI: 10.7554/eLife.71900.
[6]FALKOWSKI P G, FENCHEL T, DELONG E F. Themicrobial engines that drive earth’s biogeochemical cycles[J]. Science, 2008, 320(5879): 1034. DOI: 10.1126/science.1153213.
[7]DELGADO-BAQUERIZO M, REICH P B, TRIVEDI C,et al. Multiple elements of soil biodiversity drive ecosystemfunctions across biomes[J]. Nature Ecology amp; Evolution,2020, 4(2): 210. DOI: 10.1038/s41559-019-1084-y.
[8]SAVARY S, WILLOCQUET L, PETHYBRIDGE S J, etal. The global burden of pathogens and pests on majorfood crops[J]. Nature Ecology amp; Evolution, 2019, 3(3):430. DOI: 10.1038/s41559-018-0793-y.
[9]YANG J H, LIU H X, ZHU G M, et al. Diversity analysisof antagonists from rice-associated bacteria and theirapplication in biocontrol of rice diseases[J]. Journal ofApplied Microbiology, 2008, 104(1): 91. DOI: 10.1111/j.1365-2672.2007.03534.x.
[10]BULL C T, DE BOER S H, DENNY T P, et al. Comprehensivelist of names of plant pathogenic bacteria, 1980-2007[J]. Journal of Plant Pathology, 2010, 92(3): 551.DOI: 10.4454/jpp.v92i3.302.
[11]WANNER L A, KIRK W W, QU X S. Field efficacy ofnonpathogenic Streptomyces species against potato commonscab[J]. Journal of Applied Microbiology, 2014,116(1): 123. DOI: 10.1111/jam.12336.
[12]NICHOLLS C, ALTIERI M. Agroecology: principles forthe conversion and redesign of farming systems[J]. Journalof Ecosystem and Ecography, 2016, S5: 010. DOI:10.4172/2157-7625.S5-010.
[13]YU R P, YANG H, XING Y, et al. Belowground processesand sustainability in agroecosystems with intercropping[J]. Plant and Soil, 2022, 476(1): 263. DOI: 10.1007/s11104-022-05487-1.
[14]YANG J M, DUAN Y J, LIU X Y, et al. Reduction ofbanana fusarium wilt associated with soil microbiomereconstruction through green manure intercropping[J].Agriculture, Ecosystems amp; Environment, 2022, 337(73):108065 DOI: 10.1016/j.agee.2022.108065.
[15]LI X G, DE BOER W, ZHANG Y N, et al. Suppressionof soil-borne Fusarium pathogens of peanut by intercroppingwith the medicinal herb Atractylodes lancea[J]. SoilBiology and Biochemistry, 2018, 116: 120. DOI: 10.1016/j.soilbio.2017.09.029.
[16]LARKIN R P, HONEYCUTT C W, GRIFFIN T S, et al.Effects of different potato cropping system approaches and water management on soilborne diseases and soilmicrobial communities[J]. Phytopathology, 2010, 101(1):58. DOI: 10.1094/PHYTO-04-10-0100.
[17]GUO Z B, WAN S X, HUA K K, et al. Fertilization regimehas a greater effect on soil microbial communitystructure than crop rotation and growth stage in an agroecosystem[J]. Applied Soil Ecology, 2020, 149(1): 103510.DOI: 10.1016/j.apsoil.2020.103510.
[18]JIANG Y L, ZHANG J, MANUEL D, et al. Rotationcropping and organic fertilizer jointly promote soil healthand crop production[J]. Journal of Environmental Management,2022, 315: 115190. DOI: 10.1016/j.jenvman.2022.115190.
[19]WANG G Z, LI X G, XI X Q, et al. Crop diversificationreinforces soil microbiome functions and soil health[J].Plant and Soil, 2022, 476(1): 375. DOI: 10.1007/s11104-022-05436-y.
[20]KOWALCHUK G A, BUMA D S, DE BOER W, et al.Effects of above-ground plant species composition anddiversity on the diversity of soil-borne microorganisms[J].Antonie van Leeuwenhoek, 2002, 81(1): 509. DOI: 10.1023/A:1020565523615.
[21]GUSEVA K, DARCY S, SIMON E, et al. From diversityto complexity: microbial networks in soils[J]. SoilBiology and Biochemistry, 2022, 169: 108604. DOI: 10.1016/j.soilbio.2022.108604.
[22]DENG Y, JIANG Y H, YANG Y, et al. Molecular ecologicalnetwork analyses[J]. BMC Bioinformatics, 2012,13(1): 113. DOI: 10.1186/1471-2105-13-113.
[23]KURTZ Z D, MÜLLER C L, MIRALDI E R, et al.Sparse and compositionally robust inference of microbialecological networks[J]. PLoS Computational Biology,2015, 11(5): e1004226. DOI: 10.1371/journal.pcbi.1004226.
[24]GUO S, TAO C Y, JOUSSET A, et al. Trophic interactionsbetween predatory protists and pathogen-suppressivebacteria impact plant health[J]. The ISME Journal,2022, 16(8): 1932. DOI: 10.1038/s41396-022-01244-5.
[25]CALLAHAN B J, MCMURDIE P J, HOLMES S P. Exactsequence variants should replace operational taxonomicunits in marker-gene data analysis[J]. The ISME Journal,2017, 11(12): 2639. DOI: 10.1038/ismej.2017.119.
[26]HUNJAN M S, SABHIKHI H S. Designing a crop rotationstrategy to manage Streptomyces scabies causingpotato scab in north India[J]. Journal of Phytopathology,2020, 168(7/8): 469. DOI: 10.1111/jph.12911.
[27]STEWART E J. Growing unculturable bacteria[J]. Journalof Bacteriology, 2012, 194(16): 4151. DOI: 10.1128/jb.00345-12.
[28]EDGAR R C. Search and clustering orders of magnitudefaster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460.DOI: 10.1093/bioinformatics/btq461.
[29]ROGNES T, FLOURI T, NICHOLS B, et al. VSEARCH:a versatile open source tool for metagenomics[J]. PeerJ,2016, 4: e2584. DOI: 10.7717/peerj.2584.
[30]LIU Y X, CHEN L, MA T F, et al. EasyAmplicon: aneasy-to-use, open-source, reproducible, and communitybasedpipeline for amplicon data analysis in microbiome research[J]. iMeta, 2023, 2(1): e83. DOI: 10.1002/imt2.83.
[31]NEWMAN M E J. Modularity and community structurein networks[J]. Proceedings of the National Academy ofSciences, 2006, 103(23): 8577. DOI: 10.1073/pnas.0601602103.
[32]OLESEN J M, BASCOMPTE J, DUPONT Y L, et al.The modularity of pollination networks[J]. Proceedingsof the National Academy of Sciences, 2007, 104(50):19891. DOI: 10.1073/pnas.0706375104.
[33]LOUCA S, PARFREY L W, DOEBELI M. Decouplingfunction and taxonomy in the global ocean microbiome[J]. Science, 2016, 353(6305): 1272. DOI: 10.1126/science.aaf4507.
[34]GODFRAY H C J, BEDDINGTON J R, CRUTE I R,et al. Food security: the challenge of feeding 9 billion people[J]. Science, 2010, 327(5967): 812. DOI: 10.1126/science.118538.
[35]TITTONELL P. Ecological intensification of agriculturesustainableby nature[J]. Current Opinion in EnvironmentalSustainability, 2014, 8: 53. DOI: 10.1016/j.cosust.2014.08.006.
[36]QIN S H, YEBOAH S, CAO L, et al. Breaking continuouspotato cropping with legumes improves soil microbialcommunities, enzyme activities and tuber yield[J].PLoS One, 2017, 12(5): e0175934. DOI: 10.1371/journal.pone.0175934.
[37]SHI Y, DELGADO-BAQUERIZO M, LI Y T, et al.Abundance of kinless hubs within soil microbial networksare associated with high functional potential in agriculturalecosystems[J]. Environment International, 2020,142: 105869. DOI: 10.1016/j.envint.2020.105869.
[38]ZHOU Z B, ZHANG Y J, ZHANG F G. Abundant andrare bacteria possess different diversity and function incrop monoculture and rotation systems across regionalfarmland[J]. Soil Biology and Biochemistry, 2022, 171:108742. DOI: 10.1016/j.soilbio.2022.108742.
[39]TIEMANN L K, GRANDY A S, ATKINSON E E, et al.Crop rotational diversity enhances belowground communitiesand functions in an agroecosystem[J]. Ecology Letters,2015, 18(8): 761. DOI: 10.1111/ele.12453.
[40]TOJU H, PEAY K G, YAMAMICHI M, et al. Core microbiomesfor sustainable agroecosystems[J]. Nature Plants,2018, 4(5): 247. DOI: 10.1038/s41477-018-0139-4.
[41]FAN K K, DELGADO-BAQUERIZO M, GUO X S, etal. Biodiversity of key-stone phylotypes determines cropproduction in a 4-decade fertilization experiment[J]. TheISME Journal, 2021, 15(2): 550. DOI: 10.1038/s41396-020-00796-8.
[42]ZHENG H P, YANG T J, BAO Y Z, et al. Network analysisand subsequent culturing reveal keystone taxa involvedin microbial litter decomposition dynamics[J].Soil Biology and Biochemistry, 2021, 157(1): 108230.DOI: 10.1016/j.soilbio.2021.108230.
[43]YUAN M M, GUO X, WU L W, et al. Climate warmingenhances microbial network complexity and stability[J].Nature Climate Change, 2021, 11(4): 343. DOI: 10.1038/s41558-021-00989-9.
[44]BEI Q C, MOSER G, MÜLLER C, et al. Seasonality affectsfunction and complexity but not diversity of therhizosphere microbiome in European temperate grassland[J]. Science of the Total Environment, 2021, 784:147036. DOI: 10.1016/j.scitotenv.2021.147036.
[45]RAAIJMAKERS J M, MAZZOLA M. Soil immune responses[J]. Science, 2016, 352(6292): 1392. DOI: 10.1126/science.aaf3252.
[46]ZHANG Y C, JIANG G D, DING Y S, et al. Geneticbackground affects pathogenicity island function and pathogenemergence in Streptomyces[J]. Molecular PlantPathology, 2018, 19(7): 1733. DOI: 10.1111/mpp.12656.
[47]BLANCHET F G, CAZELLES K, GRAVEL D. Co-occurrenceis not evidence of ecological interactions[J].Ecology Letters, 2020, 23(7): 1050. DOI: 10.1111/ele.13525.
[48]LI P F, LIU J, SALEEM M, et al. Reduced chemodiversitysuppresses rhizosphere microbiome functioningin the mono-cropped agroecosystems[J]. Microbiome,2022, 10(1): 108. DOI: 10.1186/s40168-022-01287-y.
[49]WANG T, YANG K X, MA Q Y, et al. Rhizosphere microbialcommunity diversity and function analysis of cut chrysanthemumduring continuous monocropping[J]. Frontiersin Microbiology, 2022, 13: 801546. DOI: 10.3389/fmicb.2022.801546.
[50]SUBBANNA A R N S, RAJASEKHARA H, STANLEYJ, et al. Pesticidal prospectives of chitinolytic bacteriain agricultural pest management[J]. Soil Biologyand Biochemistry, 2018, 116: 52. DOI: 10.1016/j.soilbio.2017.09.019.
[51]PASCUAL J, BLANCO S, RAMOS J L, et al. Responsesof bulk and rhizosphere soil microbial communitiesto thermoclimatic changes in a Mediterranean ecosystem[J]. Soil Biology and Biochemistry, 2018, 118:130. DOI: 10.1016/j.soilbio.2017.12.013.
[52]BROWN R W, CHADWICK D R, BENDING G D, etal. Nutrient (C, N and P) enrichment induces significantchanges in the soil metabolite profile and microbial carbonpartitioning[J]. Soil Biology and Biochemistry, 2022,172: 108779. DOI: 10.1016/j.soilbio.2022.108779.
[53]LI B B, ROLEY S S, DUNCAN D S, et al. Long-termexcess nitrogen fertilizer increases sensitivity of soil microbialcommunity to seasonal change revealed by ecologicalnetwork and metagenome analyses[J]. Soil Biologyand Biochemistry, 2021, 160: 108349. DOI: 10.1016/j.soilbio.2021.108349.
责任编辑:何謦成
基金项目:国家马铃薯产业体系(CARS-09-15P)。
猜你喜欢
幼儿100(2023年39期)2023-10-23 11:36:32
青少年科技博览(中学版)(2022年6期)2022-12-27 19:44:27
中国土壤与肥料(2021年5期)2021-12-12 02:02:11
今日农业(2021年21期)2021-11-26 05:07:00
军事文摘(2021年22期)2021-11-26 00:43:51
今日农业(2021年14期)2021-10-14 08:35:40
金桥(2021年7期)2021-07-22 01:55:38
今日农业(2020年20期)2020-11-26 06:09:10
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
