
骨质疏松中衰老相关分泌表型调控机制的研究进展
2024-07-11 10:19:33杨超富谭国庆徐展望
中国全科医学 2024年29期
杨超富 谭国庆 徐展望
基金项目:国家自然科学基金面上项目(82174410);山东省自然科学基金资助项目(ZR2020KH011);山东省自然科学基金面上项目(ZR2020MH362);全国名老中医专家传承工作室建设项目(国中医药人教函[202275号])
引用本文:杨超富,谭国庆,徐展望.骨质疏松中衰老相关分泌表型调控机制的研究进展[J]. 中国全科医学,2024,27(29):3685-3695. DOI:10.12114/j.issn.1007-9572.2023.0721. [www.chinagp.net]
YANG C F,TAN G Q,XU Z W. Research progress on the regulation mechanism of aging-related secretory phenotype in osteoporosis[J]. Chinese General Practice,2024,27(29):3685-3695.
? Editorial Office of Chinese General Practice. This is an open access article under the CC BY-NC-ND 4.0 license.
【摘要】 衰老相关分泌表型(SASP)是细胞衰老的重要特征,在调控疾病微环境中具有重要作用。目前,对于SASP干预骨代谢、诱导骨流失的作用机制了解有限,因此,本文探讨了在骨质疏松模型中SASP的调控机制,并归纳总结了其调控特点:SASP在衰老骨细胞中充分表达,以自分泌/旁分泌的方式将衰老效应传递到间充质干细胞,从而干预其成骨分化;SASP激活免疫细胞,并促进其衰老,从而诱导炎性组织微环境的形成,加重骨流失;线粒体稳态失调、病理性血糖升高、肥胖诱导的脂肪蓄积均会促进SASP的表达,从而扰乱微环境稳态,将衰老效应传递到骨组织。所以,有必要深入了解SASP在骨质疏松中的作用,为后续开发抗SASP疗法治疗骨质疏松提供借鉴。
【关键词】 骨质疏松;衰老相关分泌表型;细胞衰老;代谢紊乱;免疫调节
【中图分类号】 R 681 【文献标识码】 A DOI:10.12114/j.issn.1007-9572.2023.0721
Research Progress on the Regulation Mechanism of Senescence-associated Secretory Phenotype in Osteoporosis
YANG Chaofu1,TAN Guoqing2*,XU Zhanwang2
1.Shandong University of Traditional Chinese Medicine,Jinan 250039,China
2.Spinal Department,Affiliated Hospital of Shandong University of Traditional Chinese Medicine,Jinan 250013,China
*Corresponding author:TAN Guoqing,Associate chief physician/Associate professor;E-mail:yxkmt@hotmail.com
【Abstract】 Senescence-associated secretory phenotype(SASP) is an important feature of cellular senescence and plays an important role in regulating the disease microenvironment. At present,the role of SASP in intervening bone metabolism and inducing bone loss is very limited. Therefore,this paper discusses the regulatory mechanism of SASP in osteoporosis models and summarizes its regulatory characteristics:SASP is fully expressed in senescent bone cells and transmits aging effects to mesenchymal stem cells in an autocrine/paracrine manner,thereby interfering with osteogenic differentiation. SASP activates immune cells and promotes their aging,thus inducing the formation of inflammatory tissue microenvironment and aggravating bone loss. Mitochondrial homeostasis,pathologic hyperglycemia,and obesity-induced fat accumulation all promote SASP expression,thus disrupting microenvironmental homeostasis and transmitting aging effects to bone tissue. To sum up,understanding the role of SASP in osteoporosis lays a solid foundation for us to develop anti-SASP therapy for osteoporosis in the future.
【Key words】 Osteoporosis;Senescence-associated secretory phenotype;Cell senescence;Metabolic disorders;Immunomodulation
骨质疏松症是一种因骨密度下降导致骨折风险增高的全身性代谢骨病。2021年,一项全国性、多中心的双能X线吸收测定法(DXA)研究调查了中国40岁以上人群骨质疏松症的标准化患病率,该项研究显示:40岁及以上女性骨质疏松症的总体患病率为20.6%,男性为5.0%。绝经后女性骨质疏松症患病率为32.1%,50岁及以上男性骨质疏松症患病率为6.9%[1]。虽然该结果与其他相关研究[2]均表明,女性比男性更容易患上骨质疏松症和随后的骨折,但对于40岁及以上的男性来说,其椎体骨折发生率要高于女性。因此,对于不同性别的骨质疏松症患者来说,降低脆性骨折发生风险,增强骨密度和骨稳定性,是治疗的重中之重。
衰老相关分泌表型(SASP)的概念最早由Jean-Philippe Copper于2008年提出,用于人类恶性肿瘤的研究。这项研究表明,衰老细胞可以通过分泌一些物质促进癌前细胞癌变,这些物质统称为SASP因子[3]。随着不断研究,SASP因子家族也在发展壮大;SASP由一系列促炎因子、趋化因子、生长因子和蛋白酶组成,是细胞衰老时体内外多种因子刺激产生的,包括肿瘤坏死因子(TNF)-α、白介素(IL)-6、IL-1、IL-8、基质金属蛋白酶(MMP)、粒细胞集落刺激因子(G-CSF)等。而近年来SASP在调控疾病微环境中的作用越来越得到重视;SASP能影响细胞行为,并与细胞应激性衰老、年龄相关老化有着千丝万缕的联系,而这种联系很大程度上在于SASP可以激活免疫系统,并促进慢性炎症的形成[4-5]。本文旨在利用SASP相关观点阐述骨质疏松的形成机制,并重点探讨了骨骼老化如何驱动成骨能力降低、SASP如何激活免疫应答以及代谢紊乱如何诱发SASP等重点问题。
本文文献检索策略:计算机检索PubMed、Web of Science等数据库,将检索时间设定为建库至2023年7月,检索词包括“osteoporosis”“senescence-associated secretory phenotype”“Bone loss”“cell aging”“metabolic disorders”等。纳入标准:内容涉及以上关键词的文献,优先选择高质量期刊的文献。排除标准:文献内容与本文主题无关联、文章质量较差、无法获得全文的文献。
1 SASP:干预间充质干细胞(MSC)成骨分化,调控骨质疏松中组织的再生与修复
对于骨质疏松症患者的监管,基于SASP的观点认为:年龄相关的骨流失只是诱发骨形成减少的一个重要因素,但不是全部——持续的DNA损伤及创伤后机体的应激反应会导致大量SASP因子释放,从而介导全身慢性无菌性炎症,也会显著影响骨代谢平衡,尤其是DNA损伤导致的永久性细胞周期停滞,会影响MSC活性以及增殖分化能力,这会对骨组织的再生与修复造成不可逆的损伤。因此,利用SASP调控MSC使之具有稳定的增殖分化能力,是其成骨分化的基础。
骨微环境中SASP因子的释放是影响MSC活性的重要因素。一项研究表明,衰老的骨细胞通过旁分泌途径改善骨髓间充质干细胞(BMSCs)的分化潜力,其过程表现为:(1)应激来源激活导致骨细胞核内染色质结构的破坏、核完整性的破坏以及衰老相关异染色质灶(SAHF)的形成,并导致γ-H2AX(一种DNA双链断裂标志物)积累。(2)衰老骨细胞失去分裂能力并经历生长停滞,但其仍具有代谢活性并分泌SASP,导致IL-6、IL-1α、MMP-3和抵抗素等细胞因子积累。(3)BMSCs集落形成的能力被轻度破坏,成骨、成脂分化潜力降低。(4)经体外诱导的BMSCs矿化结节形成能力显著降低,脂肪形成能力显著增加,导致骨骼老化与骨质流失[6]。
骨稳态的调节依赖MSC和造血干细胞(HSC)谱系之间的相互作用,且在衰老进程中尤为明显。在衰老过程中,这些细胞谱系发生剧烈变化,导致骨髓-淋巴造血和脂肪-成骨分化之间的不平衡[7-8],这导致骨髓生成和脂肪生成增加,而不是淋巴细胞生成和骨生成。SASP观点认为:衰老及数量减少的骨细胞会产生SASP因子,定向改变MSC谱系,导致破骨细胞增加。该过程具体表现为:(1)MSC、HSC多次传代导致细胞衰老,并分泌SASP,越来越多的SASP因子(TNF-α、IL-1β和IL-6等)参与循环,骨髓成熟度增加,随着基因表达产物增多,诱导衰老进行性加重,导致骨细胞出现衰老表型甚至消融,引起数目减少。(2)骨细胞分泌SASP因子,反向作用MSC,下调成骨通路Wnt、Hedgehog、Notch表达,并导致MSC成脂分化增加,脂肪积累。(3)循环性RANKL增加,循环性骨保护素(OPG)减少,RANKL/OPG比率下降、血清Ⅰ型c-端肽(CTX)分泌增加,破骨细胞生成增加,导致骨质减少[9]。
诸如衰老的骨细胞等可以通过产生SASP来诱导MSC的衰老,因此要稳定应用MSC等再生医学成果来改善骨质流失、骨缺损的情况,则需要探讨MSC传代引发自身衰老导致的问题:许多研究证实,传代晚期MSC会分泌SASP因子促进早期MSC衰老,极大影响了利用干细胞移植促进组织再生的策略[10-11]。且传代晚期MSC的成骨、成脂分化潜力减弱,其稳态的维持却依赖于产生的SASP:含有脂质代谢物的细胞外囊泡经由衰老MSC产生、并具有诱导细胞衰老和凋亡的毒性,反而加重了衰老进程[12]。小尺寸的MSC具有更高的生长潜力和更低的衰老率[13],SASP通过自分泌/旁分泌正调节环路维持衰老进程[14]。相关研究证实,脐带血来源的间充质干细胞(UCB-MSC)的小细胞具有更强的增殖能力,并且表现出更低的细胞衰老[15]:UCB-MSC小细胞多次传代产生低剂量的以生长调节致癌基因α、IL-8为主的SASP因子,其与趋化因子受体2(CXCR2)结合加速细胞衰老进程,并受Toll样受体2(TLR2)和TLR5促进、MSC小细胞分泌的si-RNA抑制。传代晚期MSC细胞可通过旁分泌方式释放IL-1α、IL-8等炎性因子以核因子(NF)-κB依赖的方式诱导早期MSC的衰老[16]。信号蛋白WNT3可通过抑制SASP因子的旁分泌途径来中断MSC的衰老进程:并且WNT信号不是通过调节增殖和分化,而是通过保护细胞免受衰老的有害影响来支持MSC增殖和发育潜力[17]。MSC中转录因子TWIST1的沉默会增加衰老的发生,并导致其代谢异常,具体可表现为细胞耗氧率增加[18]。核层缺陷的发生是导致MSC早衰的原因之一:异常的前核纤层蛋白A通过MSC中的GATA4依赖性途径触发旁分泌衰老;GATA4的缺失通过早老蛋白或前核纤层蛋白A抑制MSC中的NF-κB和MCP-1消除了SASP依赖性衰老[19]。BMI-1是调节MSC自我更新的重要因子,衰老MSC细胞中SASP因子IL-1α升高,导致B细胞特异性莫洛尼鼠白血病病毒整合位点1 BMI-1表达降低,是MSC早衰的原因之一[20]。
mi-RNA被认为是调控MSC行为的重要靶标,包封mi-RNA的小细胞外囊泡可作为无细胞疗法应用于骨再生与修复。SASP观点认为:mi-RNA借由外泌体囊泡运输,通过旁分泌途径诱导MSC出现衰老表型。此外mi-RNA也可由MSC产生,通过调节特定的靶基因加速衰老进程。miR-29c-3p可通过p53-p21和p16-pRB途径靶向CNOT6促进MSC衰老[21]。miR-31在老年人和骨质疏松症患者的血浆中升高,经由衰老来源细胞的外泌体囊泡运输,并被MSC吸收,通过敲低其靶标frizzed-3来抑制成骨分化[22]。miR-335可显著调节MSC的增殖和分化,在体内通过响应诱导细胞衰老的刺激而增加。miR-335的过表达借由外泌体囊泡运输作用于MSC,并降低其软骨成骨潜力[23]。P65可以防止MSC产生SASP因子,并防止MSC之间的旁分泌衰老和通过小细胞外囊泡的促炎信息的传递[24]。
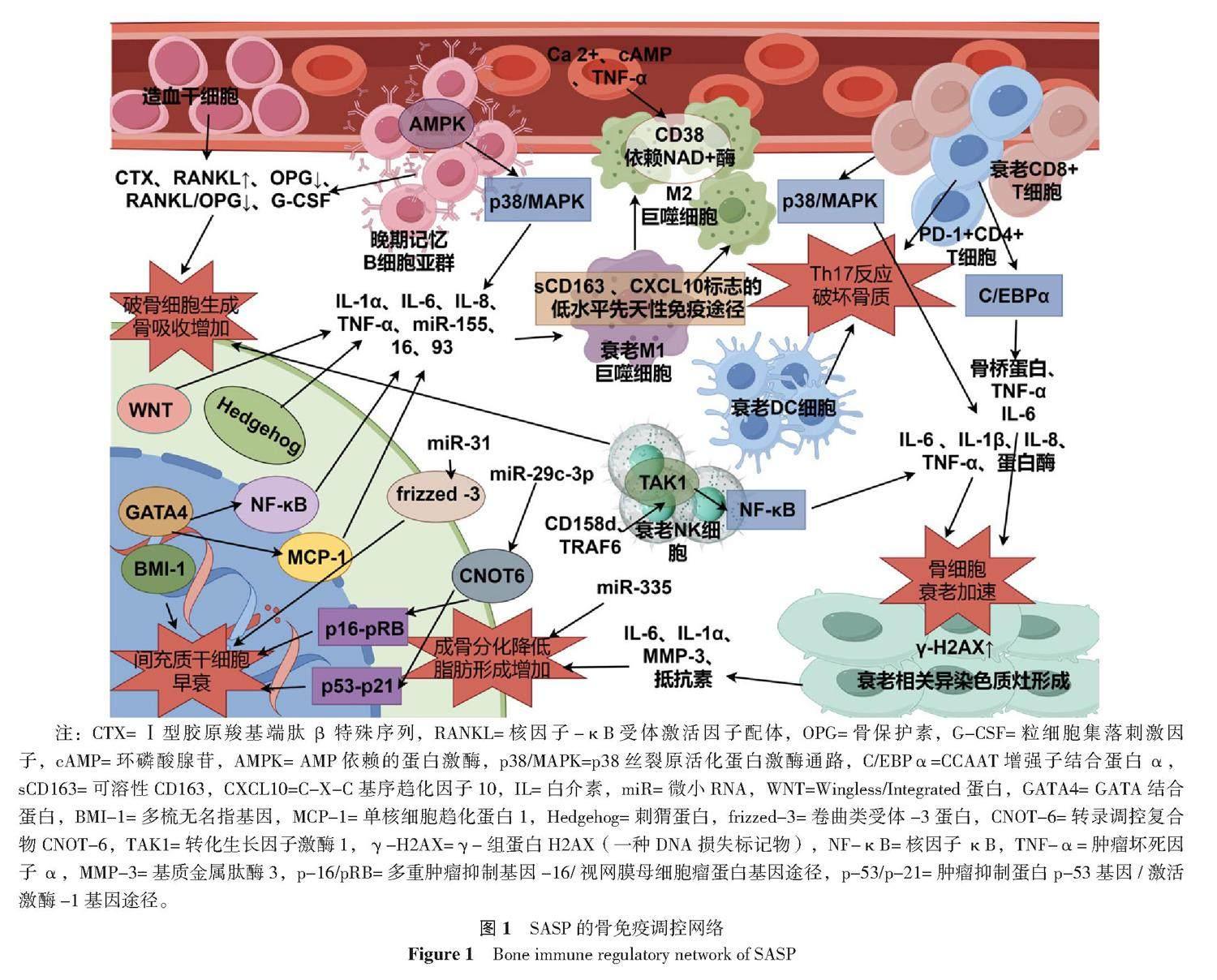
2 SASP:介导骨质疏松中慢性炎症反应,调控骨免疫网络
骨质疏松症患者的骨微环境中含有大量促炎因子,可介导全身出现慢性炎性反应。SASP观点认为:这种慢性炎症状态的建立会显著加重细胞衰老,促炎因子是SASP的重要组成部分可由全身出现衰老表型的细胞释放,而抵抗这种炎性状态很大程度上依赖免疫系统,尤其是免疫细胞的行为。该笔者以SASP因子作为桥梁,探讨了免疫系统与骨代谢平衡之间的联系以及衰老的免疫细胞出现的变化。建立骨免疫调控机制,探讨如何通过免疫途径改善骨质疏松症患者促炎状态导致的骨
流失。
2.1 巨噬细胞极化与SASP
在衰老微环境中,巨噬细胞可以被多种细胞因子募集,包括SASP因子和NK细胞分泌组,由此产生免疫功能上的联系。NK细胞通过与衰老细胞相互作用产生干扰素γ(IFN-γ)[25-26],招募巨噬细胞。此外,巨噬细胞也响应SASP因子CCL2、CXCL1、CXCL16和IL-8等的募集[27],SASP相关的CCL2导致促炎性M1巨噬细胞的积累[28-29]。且随着衰老进行性加重,巨噬细胞表现出高炎性、低免疫活性状态,并出现衰老特征,其具体过程为:(1)巨噬细胞响应年龄衰老,激活sCD163、CXCL10标志的低水平先天性免疫途径,导致功能失调[30]。(2)SASP因子释放作用于巨噬细胞,导致TNF-α水平增加。(3)巨噬细胞出现衰老特征,并向M2型转变,并下调IL-10水平、显著降低吞噬能力[31]。
衰老细胞难以被巨噬细胞杀死,并且可抑制巨噬细胞通过旁分泌途径识别SASP信号清除凋亡细胞残体的能力。衰老细胞介导的胞吞作用抑制(SCES)会导致巨噬细胞功能瘫痪,其原因是CD47表达增强,同时衰老细胞中CD47修饰酶QPCT/L增加。SCES通过干扰SIRPα-CD47-SHP-1轴或QPCT/L活性而抑制巨噬细胞能力。CD47和CD24表达增加是衰老细胞介导稳态功能失调(例如胞吐作用)的组成部分,而胞吐作用必须有效发生才能维持组织稳态并抑制自身免疫[32-35]。CD38是巨噬细胞功能调节的重要因子,可以调节细胞Ca2+代谢,具有抗破骨细胞生成的特性。CD38表达增强可以减少破骨细胞的数量和骨吸收;SASP因子可以诱导巨噬细胞中CD38表达,这些M1样巨噬细胞表达高水平的CD38并增强CD38依赖性NAD+酶活性,从而降低组织NAD+水平,而与衰老相关的NAD+水平的降低减少了SASP因子的产生,并减轻其病理作用[36-38]。并且,通过Ca2+、cAMP和TNF-α调节CD38的表达,有助于将破骨细胞和成骨细胞的强代谢活性与其各自的骨吸收和骨重塑功能结合起来[39-40]。
2.2 多免疫细胞串扰的促炎网络与SASP
为响应SASP因子,多种免疫细胞集合串扰产生庞杂的促炎网络,极大影响着骨微环境的稳定,往往单个免疫细胞集群没有特殊作用,而多个免疫细胞集群却受一种或几种促炎因子调节。因此,探讨多免疫细胞整体调控SASP网络的能力尤为重要。
单核细胞作为优良的细胞储备,可以充当破骨细胞、巨噬细胞、树突状细胞的前体[41],且可以产生趋化因子募集免疫细胞到骨重塑位点[42]。并且,单核细胞集群具有异质性,根据CD14、CD16表达量的不同分为3个子集:经典(CD14高/CD16-)、中间(CD14高/CD16+)和非经典(CD14低/CD16+)。SASP因子诱导了非经典子集CD16+表现出最明显的促炎状态、高miR-146a(一种负向调节TLR 通路的mi-RNA)表达,这被认为是单核细胞的衰老状态。此外,NF-κB和 IL-1α可能是介导单核细胞出现衰老表型的关键靶点,血浆环境中TNF-α、IL-8水平升高则会导致这种衰老表型进行性加深[43]。此外,又有研究发现,SASP因子GDF-15可以诱导单核细胞产生更多CD16+表型,并且可以通过抑制其线粒体呼吸的能力,促进衰老[44]。树突状细胞(DC细胞)在维持免疫稳态中尤为重要,未成熟DC细胞可以诱导T细胞反应缺失及调节性T细胞(Treg)反应来促进免疫耐受,从而终止炎症[45],并减轻炎症性的骨流失[46]。而SASP因子大量释放导致的免疫微环境失调会刺激DC细胞成熟——成熟的DC细胞反而会抑制Treg反应,并产生免疫刺激性T细胞(例如Th17)反应破坏骨质[47]。不止于此,DC细胞介导的具有免疫效应的外泌体囊泡也参与调控SASP:在响应感染、促炎因子等信号后,DC细胞触发炎性小体活化[48],并分泌含有IL-1B等促炎因子的外泌体[49],借由旁分泌途径将衰老效应传递给周围细胞。自然杀伤细胞(NK细胞)可以通过生成M-CSF 和 RANKL诱导单核细胞分化为破骨细胞,从而加剧骨流失,而其本身的破骨细胞生成能力较差[50]。SASP观点认为:衰老细胞可分泌多种趋化因子(CXCL10等)通过与CXCR3结合增强NK细胞增殖迁移的能力,从而介导其对衰老细胞的清除[51]。并且CD158d的表达可以刺激静息NK细胞,通过募集TRAF6激活 TAK1 来诱导NF-κB信号产生[52],从而导致NK细胞衰老并出现SASP[53],该分泌组可以显著促进血管生成。
T细胞可以响应微环境刺激并做出其他免疫细胞比拟不了的精细化反应,在骨平衡中,不同亚群的T细胞有着极为重要的作用:T细胞功能的实现有赖于CD4+T和CD8+T细胞亚群的协同效应。其中,由CD4+T分化来的Th17主要负责刺激破骨细胞产生,从而介导骨吸收过程[54-55],而Treg则能有效抑制骨吸收,二者的动态平衡是维持骨代谢稳定的关键部分。不止于此,CD8+T细胞可通过分泌骨保护素(OPG)[56]和IFN-γ[57]抑制破骨细胞生成。SASP观点认为:两种T细胞亚群会出现衰老表型,并接受SASP因子的调控,表现出代谢活跃的高促炎状态,并显著加强骨流失。衰老CD4+T表现出PD-1+记忆表型——这些细胞在T细胞受体刺激下不会增殖,并产生大量SASP因子:例如骨桥蛋白、TNF-α和IL-6[58]等,这与C/EBPα表达上调有关。不同于衰老CD4+T,衰老的CD8+ T有自身独特的SASP(以CD8+CD45RA+CD27-T子群最具异质性):即产生更多的IL-6、IL-1β,并分泌蛋白酶(组织蛋白酶和丝氨酸蛋白酶,还包括ADAM家族和金属蛋白酶),并受p38/MAPK调控[59]。
在B细胞中,只有记忆B细胞亚群会表达SASP,尤其是晚期/耗尽记忆B细胞(LM B细胞)。LM B细胞亚群通过自发激活AMPK活性,诱导p38/MAPK表达,并导致促炎因子(TNF-α、IL-6、IL-8等)、炎性mi-RNA ( miR-155、16、93等)释放,通过旁分泌途径将衰老信号传递到周围组织[54]。SASP介导的高促炎状态刺激B细胞产生RANKL和粒细胞集落刺激因子(G-CSF)[60-61],从而激活了破骨细胞的生成,将B细胞促进骨重塑的作用转化为骨吸收[62]。具体机制见图1。
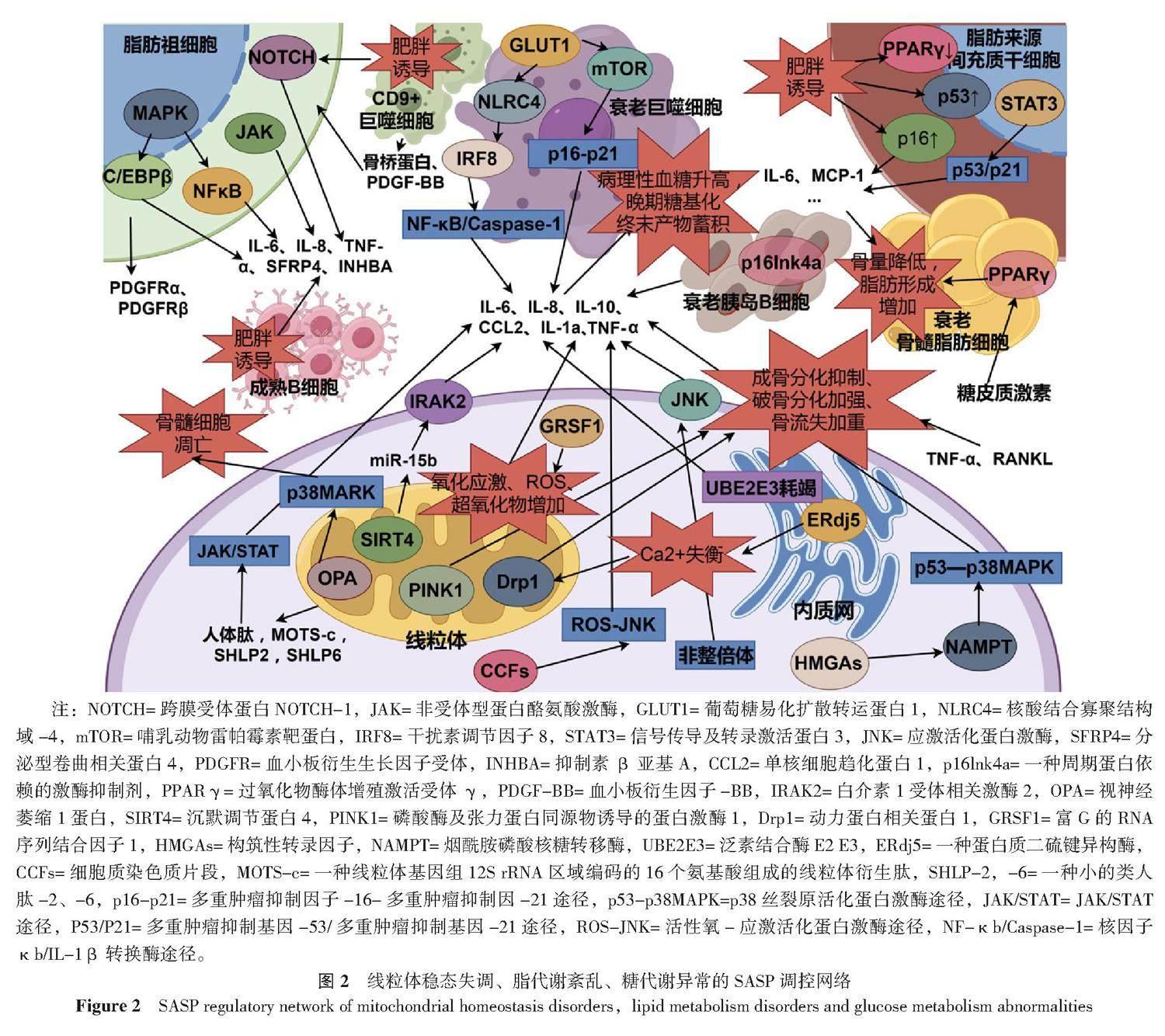
3 SASP:调控骨质疏松中多重代谢反应
骨质疏松症患者的一个显著特征是全身多种代谢失调,SASP被认为可以介导代谢失常后的二次损害,并与不良组织微环境的形成有关。在本文这一部分笔者探讨了部分代谢失调与SASP的调控之间的关系,以及他们可能对骨代谢平衡造成的危害,以期构建更为全面详细的SASP控制网络。
3.1 线粒体稳态失调:能量代谢异常、氧化应激与SASP
线粒体稳态对维持细胞能量供应以及介导全身的抗氧化代谢极其重要,换言之,线粒体稳态失调会导致能量代谢异常,并使细胞出现氧化应激状态,这是触发SASP的重要条件。
衰老会导致线粒体功能障碍(SAMD),并促进SASP产生。沉默调节蛋白4(SIRT4)仅在线粒体中表达,并受miR-15b负向调控。在衰老细胞中SIRT4表达上升,导致miR-15b生成受抑制。靶向抑制miR-15b增强SIRT4表达会促进线粒体内活性氧(ROS)生成,并降低线粒体膜电位——导致线粒体功能紊乱。并且,抑制miR-15b会导致SASP以SIRT4依赖的方式释放,并阻止了正常水平下miR-15b以IRAK2依赖方式抑制SASP因子IL-6、IL-8的产生[63-64]。GRSF1 是维持线粒体氧化磷酸化所必需的蛋白,衰老细胞中的 GRSF1 水平因蛋白质稳定性降低而下降,GRSF1水平降低会导致线粒体应激,并导致超氧化物生成增加、DNA损伤灶增加和细胞增殖减少,并导致细胞出现衰老表型:即衰老相关β-半乳糖苷酶 (SA-β-gal) 活性上升、SASP因子IL-6的产生和分泌[65]。值得一提的是,SAMD诱导的ROS可调控SASP的产生,但SASP不可以反向介导SAMD,SASP会以旁分泌的方式将衰老效应传递到周围更多组织中[66]。这表明,对于SAMD-ROS效应诱导的衰老,无法通过抑制SASP因子的产生来控制,抗氧化以及增强线粒体活性的药物可能是对抗该效应的
良策。
衰老导致线粒体呼吸能力减弱,使细胞转向主要依靠糖酵解来供能,并伴随SASP产生。烟酰胺磷酸核糖基转移酶(NAMPT)是NAD+挽救途径的限速酶,在衰老过程中受HMGAs调控。HMGAs/NAMPT/NAD+信号轴通过增强糖酵解和线粒体呼吸调控SASP因子的释放,该过程为:HMGAs/NAMPT通过NAD+介导的AMPK激酶抑制过程(p53介导的p38MAPK途径抑制),增强NF-κb活性,从而促进SASP产生[67]。令人振奋的是,衰老线粒体可通过产生线粒体衍生肽(MDP,包含:人体肽、MOTS-c、SHLP2、SHLP6等)在一定程度上维持线粒体功能水平:衰老线粒体中人体肽和MOTS-c水平升高,MOTS-c通过提升脂肪酸氧化水平来增强线粒体呼吸能力,而人体肽通过JAK/STAT途径介导少量SASP促炎因子(主要为IL-6)释放,该SASP侧重于维持衰老状态而不是加重衰老进程[68]。
SASP的调控是多元化的,揭示了介导衰老的新机制,并伴随了线粒体功能不同程度的损伤。重组人泛素蛋白连接酶(UBE2E3)耗竭会导致衰老,并表现出独特的SASP:与线粒体功能障碍导致的衰老(MiDAS)不同,UBE2E3耗竭是一种线粒体网络维护中断的衰老方式,导致线粒体稳态失调(例如线粒体的分布、质量、对毒物易感性等),并导致IL-1β增加近6倍,而IL-10等常见SASP因子增加不甚明显,这提示UBE2E3耗竭可能耦合了其他衰老途径[69]。衰老细胞的细胞核中挤出的细胞质染色质片段(CCFs)也介导了细胞衰老:功能失调的线粒体会导致核编码线粒体氧化磷酸化基因下调,并触发ROS-JNK逆行信号通路,驱动CCF的形成,从而导致SASP因子的产生[70]。染色体数量不均(非整倍体)会以c-Jun N末端激酶 (JNK)依赖的方式介导细胞衰老,并导致ROS产生、功能失调的线粒体累积以及SASP因子的释放[71]。此外,内质网驻留的二硫键还原酶ERdj5缺失会引起细胞内Ca2+失衡,并激活Drp1(一种参与线粒体裂变的胞质GTP酶),最终导致线粒体异常断裂、细胞活力下降以及SASP因子的释放[72]。
线粒体稳态失调介导的SASP效应可通过自分泌/旁分泌途径引发骨组织广泛衰老,但更特殊的是,这种SASP效应带来的线粒体融合和分裂异常也会损害成骨能力,加强破骨细胞生成,从而加重骨质疏松的发展。在氧化应激条件下,成骨细胞中Drp1的表达及其磷酸化增加,导致线粒体出现碎片、畸形和囊泡状[73]。并且SASP因子TNF-α可诱导Drp1高表达从而引发线粒体膜电位崩溃,导致线粒体功能降低,并抑制了成骨活性[74]。此外Drp1介导的线粒体超分裂还有利于破骨细胞增殖:在炎性状态下广泛生成的RANKL可通过调控Drp1及其受体蛋白Fis1、Mid49和Mid51的表达促进破骨细胞分化,从而加重骨流失[75]。
线粒体稳态失调介导的线粒体自噬减少是加重骨流失的关键因素。在氧化应激条件下,ROS及超氧化物累积并参与破坏线粒体结构,导致线粒体膜电位变化,从而使得PINK1无法与Parkin信号耦合以清除损伤线粒体和抑制SASP效应的传导[76],并且有关研究发现PINK1在骨质疏松症患者体内表达减少,这种减少导致了骨量下降,是抑制成骨分化并加重骨流失的关键[77]。
在线粒体稳态调节中OPA发挥着极为重要的作用,并将线粒体功能与SASP效应的传导和糖代谢联系起来。OPA1以长形式(L-OPA1)和短形式 (S-OPA1)调节线粒体的分裂与融合[78],在氧化应激条件下,L-OPA1裂解为S-OPA1,并显著降低线粒体功能并诱导成骨细胞凋亡[79]。而OPA过表达会激活p38MARK通路,使线粒体ATP生成减少并促进骨髓细胞凋亡[80],从而加速骨质疏松发生。而p38MARK的激活则伴随了大量SASP的释放,从而在骨组织扩大了这种效应。并且,在病理性高血糖状态下,晚期糖基化终末产物(AGE)累积,从而增强了S-OPA1并抑制L-OPA1表达、加快ROS的生成并诱导了成骨细胞凋亡[81],从而将异常的糖代谢、线粒体稳态失调与骨质疏松联系起来。
3.2 葡萄糖代谢异常与SASP
糖代谢异常介导血糖病理性升高以及晚期糖基化终末产物蓄积,这极大危害了骨骼系统的稳定。SASP观点认为:高血糖状态介导慢性炎症发生,诱发细胞衰老,并产生大量促炎性SASP因子,在炎性骨微环境下,骨代谢由骨生成转向骨吸收。
胰岛B细胞衰老诱导病理性血糖升高、并产生大量SASP因子,从而以高糖、高炎刺激破坏成骨微环境。衰老胰岛B细胞分泌CCL2、IL-1a、IL-6和TNF-α等核心SASP因子,并通过旁分泌途径诱导相邻未衰老胰岛B细胞Cdkn2a基因表达,从而导致广泛的胰岛B细胞耗竭[82]——胰岛B细胞质量降低导致胰岛素分泌不足,从而加重了高血糖状态。值得注意的是,衰老胰岛B细胞这种旁分泌效应也会扰乱胰岛A细胞功能,导致胰高血糖素分泌障碍,进而减少胰岛素的分泌[83]。衰老会损害成体胰岛B细胞的增殖和响应生长刺激的能力,p16Ink4a表达增强启动了胰岛B细胞的衰老进程,并降低其增殖能力、导致SASP因子大量释放[84]。
病理性高血糖状态会诱发低度炎症,内皮细胞和巨噬细胞是介导SASP效应的主要传播细胞,并在糖尿病性低度炎症的传播中起着极为重要的作用[85]。此外,又有研究表明:GLUT1可作为巨噬细胞中具有代表性和促进性的葡萄糖转运蛋白,并与SASP效应的介导密不可分:在高葡萄糖环境下,骨髓源性巨噬细胞(BMDM)显示出强烈的GLUT1 mRNA反应,驱动葡萄糖摄取升高,并触发mTOR磷酸化从而引发p16/p21介导的SASP因子释放[86];不止于此,高葡萄糖状态还通过NLRC4磷酸化诱导巨噬细胞衰老和SASP因子分泌,进而以IRF8依赖性途径刺激NF-κB/Caspase-1级联反应从而导致更广泛的SASP效应[87],在多重调控机制下炎性骨流失加重,衰老骨细胞得以累积。
3.3 脂质代谢紊乱与SASP
脂代谢紊乱导致脂肪蓄积,是细胞衰老进行性加重的关键因素。SASP观点认为:脂肪蓄积诱导细胞衰老并引发SASP效应,并伴随低度炎症的形成,在这种炎性组织微环境下成骨作用减弱,骨吸收增强,BMSC的成骨分化转向成脂分化。
衰老细胞在肥胖患者脂肪组织中累积,并介导SASP效应和炎症的形成。肥胖个体脂肪组织中记忆B细胞生成频率增加,而幼稚B细胞生成频率减少,并且成熟B细胞(记忆B)表现出高代谢活性并伴随IL-6、IL-8、TNF-α等大量炎性SASP因子释放,从而加重了全身炎性状态[88]。肥胖状态也诱导了衰老巨噬细胞累积:衰老巨噬细胞表现为低吞噬、高分泌活性,其中CD9+巨噬细胞通过分泌骨桥蛋白、PDGF-BB协同促进脂肪祖细胞表达PDGFRα和PDGFRβ,从而通过促进细胞外沉积以及纤维化过程来抑制脂肪组织的生成,这是难得的抗衰老过程[89]。此外,肥胖还导致衰老脂肪祖细胞累积,并激活NOTCH通路导致SASP因子SFRP4和INHBA释放,从而将脂肪生成转化为纤维生成,起到抑制肥胖的作用[90]。不仅于此,衰老的脂肪祖细胞还可通过JAK通路表达SASP,从而介导全身炎症反应[91]。此外,又有研究发现,脂肪祖细胞中SPRY1可通过抑制MAPK活性来抑制转录因子NF-κB和C/EBPβ活性从而减少SASP因子IL-6等的释放[92]。
肥胖诱导MSC出现衰老状态,并降低其增殖分化能力,这对于骨重塑是不利的。肥胖诱导脂肪来源MSC下调PPAR-γ、上调p16和p53水平,并促进炎症SASP因子IL-6、MCP-1等表达从而抑制脂肪形成能力并传递衰老效应[93]。并且,长期暴露于SASP环境中也会导致脂肪来源MSC血管生成潜力减弱,从而不利于血管化骨再生[94]。此外,又有研究将SASP因子IL-6确定为在肥胖期间诱导骨流失的关键炎症递质:IL-6通过激活BMSC中STAT3表达,并通过p53/p21途径诱导BMSC衰老,并出现SASP;利用抗体拮抗IL-6有助于维持骨髓成骨和脂肪生成之间的平衡,并抑制肥胖诱导的骨质流失中BMSC衰老[95]。而另一重要SASP因子TNF-α则主要通过将炎症效应传导到健康脂肪细胞,从而扩大衰老效应[96]。此外,肥胖介导的炎性SASP信号还可通过调控骨髓脂肪组织(BMAT)的含量来干预脂肪来源BMSC的分化进程。在肥胖诱导的骨质疏松模型中BMAT体积的增加,显著增加了骨折风险,并使BMSC由成脂分化转向成骨分化[97-98]。此外,长期施加糖皮质激素也会诱导BMAT累积:糖皮质激素通过增加15d-PGJ2等氧化脂质的合成,以激活PPAR-γ。PPAR-γ刺激关键SASP基因的表达,也促进骨髓脂肪细胞中氧化脂质的合成,形成正反馈循环,从而诱导糖皮质激素性骨质疏松的形成[99]。具体机制见图2。
4 讨论
SASP效应的发生证明了人体细胞经受超量损伤可以将其“翻译”为衰老,这种衰老效应不仅与年龄相关,还与氧化应激、炎症等组织微环境急剧变化有关,这为临床从更深刻的角度探讨骨质疏松的形成机制提供了契机。SASP相关观点认为,骨质疏松状态的形成由以下几方面因素驱动:(1)骨来源不足:与SASP效应在衰老骨组织、组织微环境、BMSC之间传递有关,其核心因素是BMSC衰老成骨能力不足。(2)骨转化抑制:与SASP效应在衰老组织、免疫细胞之间传递有关,其核心驱动因素是免疫细胞衰老形成炎性SASP加重骨流失。(3)骨代谢受损:与线粒体损伤、脂代谢紊乱(肥胖)、糖代谢失调(糖尿病病理性高血糖)介导的SASP效应在组织间传递有关,其核心因素是代谢紊乱带来的病理产物蓄积(如晚期糖基化终末产物、BMAT等)和炎性骨微环境形成有关。因此,可以利用SASP效应广泛调控骨微环境并介导炎症的特点设计抗SASP策略,对骨质疏松的治疗进行优化。
作者贡献:杨超富负责收集文献并撰写论文;谭国庆、徐展望负责对论文提供建设性意见并提供基金支持。杨超富和谭国庆对稿件整体负责。
本文无利益冲突。
参考文献
WANG L H,YU W,YIN X J,et al. Prevalence of osteoporosis and fracture in China:the China osteoporosis prevalence study[J]. JAMA Netw Open,2021,4(8):e2121106. DOI:10.1001/jamanetworkopen.2021.21106.
CONTI V,RUSSOMANNO G,CORBI G,et al. A polymorphism at the translation start site of the vitamin D receptor gene is associated with the response to anti-osteoporotic therapy in postmenopausal women from southern Italy[J]. Int J Mol Sci,2015,16(3):5452-5466. DOI:10.3390/ijms16035452.
COPP? J P,PATIL C K,RODIER F,et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor[J]. PLoS Biol,2008,6(12):2853-2868. DOI:10.1371/journal.pbio.0060301.
ZHAO R L,JIN X Y,LI A,et al. Precise diabetic wound therapy:PLS nanospheres eliminate senescent cells via DPP4 targeting and PARP1 activation[J]. Adv Sci,2022,9(1):e2104128. DOI:10.1002/advs.202104128.
JIN W N,SHI K B,HE W Y,et al. Neuroblast senescence in the aged brain augments natural killer cell cytotoxicity leading to impaired neurogenesis and cognition[J]. Nat Neurosci,2021,24(1):61-73. DOI:10.1038/s41593-020-00745-w.
HU L F,YIN C,ZHAO F,et al. Mesenchymal stem cells:cell fate decision to osteoblast or adipocyte and application in osteoporosis treatment[J]. Int J Mol Sci,2018,19(2):360. DOI:10.3390/ijms19020360.
DING P,GAO C,GAO Y S,et al. Osteocytes regulate senescence of bone and bone marrow[J]. eLife,2022,11:e81480. DOI:10.7554/eLife.81480.
SINHA S,SINHA A,DONGRE P,et al. Organelle dysfunction upon asrij depletion causes aging-like changes in mouse hematopoietic stem cells[J]. Aging Cell,2022,21(4):e13570. DOI:10.1111/acel.13570.
TRESGUERRES F G F,TORRES J,L?PEZ-QUILES J,et al. The osteocyte:a multifunctional cell within the bone[J]. Ann Anat,2020,227:151422. DOI:10.1016/j.aanat.2019.151422.
BLOCK T J,MARINKOVIC M,TRAN O N,et al. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell-based therapies[J]. Stem Cell Res Ther,2017,8(1):239. DOI:10.1186/s13287-017-0688-x.
ZHUANG Y,LI D,FU J Q,et al. Comparison of biological properties of umbilical cord-derived mesenchymal stem cells from early and late passages:immunomodulatory ability is enhanced in aged cells[J]. Mol Med Rep,2015,11(1):166-174. DOI:10.3892/mmr.2014.2755.
WANG L P,ZHANG H,XIAO X,et al. Small extracellular vesicles maintain homeostasis of senescent mesenchymal stem cells at least through excreting harmful lipids[J]. Stem Cells Dev,2023,32(17/18):565-579. DOI:10.1089/scd.2023.0079.
KIM M,BAE Y K,UM S,et al. A small-sized population of human umbilical cord blood-derived mesenchymal stem cells shows high stemness properties and therapeutic benefit[J]. Stem Cells Int,2020,2020:5924983. DOI:10.1155/2020/5924983.
LUNYAK V V,AMARO-ORTIZ A,GAUR M. Mesenchymal stem cells secretory responses:senescence messaging secretome and immunomodulation perspective[J]. Front Genet,2017,8:220. DOI:10.3389/fgene.2017.00220.
KWON J H,KIM M,UM S,et al. Senescence-associated secretory phenotype suppression mediated by small-sized mesenchymal stem cells delays cellular senescence through TLR2 and TLR5 signaling[J]. Cells,2021,10(1):63. DOI:10.3390/cells10010063.
CHOU L Y,HO C T,HUNG S C. Paracrine senescence of mesenchymal stromal cells involves inflammatory cytokines and the NF-κB pathway[J]. Cells,2022,11(20):3324. DOI:10.3390/cells11203324.
LEHMANN J,NARCISI R,FRANCESCHINI N,et al. WNT/beta-catenin signalling interrupts a senescence-induction cascade in human mesenchymal stem cells that restricts their expansion[J]. Cell Mol Life Sci,2022,79(2):82. DOI:10.1007/s00018-021-04035-x.
VOSKAMP C,ANDERSON L A,KOEVOET W J,et al. TWIST1 controls cellular senescence and energy metabolism in mesenchymal stem cells[J]. Eur Cell Mater,2021,42:401-414. DOI:10.22203/eCM.v042a25.
LEE J Y,YU K R,LEE B C,et al. GATA4-dependent regulation of the secretory phenotype via MCP-1 underlies lamin A-mediated human mesenchymal stem cell aging[J]. Exp Mol Med,2018,
50(5):1-12. DOI:10.1038/s12276-018-0092-3.
ZHENG X L,WANG Q X,XIE Z,et al. The elevated level of IL-1α in the bone marrow of aged mice leads to MSC senescence partly by down-regulating Bmi-1[J]. Exp Gerontol,2021,148:111313. DOI:10.1016/j.exger.2021.111313.
SHANG J,YAO Y,FAN X,et al. MiR-29c-3p promotes senescence of human mesenchymal stem cells by targeting CNOT6 through p53-p21 and p16-pRB pathways[J]. Biochim Biophys Acta,2016,1863(4):520-532. DOI:10.1016/j.bbamcr.2016.01.005.
WEILNER S,SCHRAML E,WIESER M,et al. Secreted microvesicular miR-31 inhibits osteogenic differentiation of mesenchymal stem cells[J]. Aging Cell,2016,15(4):744-754. DOI:10.1111/acel.12484.
TOM? M,SEP?LVEDA J C,DELGADO M,et al. MiR-335 correlates with senescence/aging in human mesenchymal stem cells and inhibits their therapeutic actions through inhibition of AP-1 activity[J]. Stem Cells,2014,32(8):2229-2244. DOI:10.1002/stem.1699.
MATO-BASALO R,MORENTE-L?PEZ M,ARNTZ O J,et al. Therapeutic potential for regulation of the nuclear factor kappa-B transcription factor p65 to prevent cellular senescence and activation of pro-inflammatory in mesenchymal stem cells[J]. Int J Mol Sci,2021,22(7):3367. DOI:10.3390/ijms22073367.
SORIANI A,IANNITTO M L,RICCI B,et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1[J]. J Immunol,2014,193(2):950-960. DOI:10.4049/jimmunol.1400271.
SHARMA C,WANG H X,LI Q L,et al. Protein acyltransferase DHHC3 regulates breast tumor growth,oxidative stress,and senescence[J]. Cancer Res,2017,77(24):6880-6890. DOI:10.1158/0008-5472.CAN-17-1536.
LEF?VRE L,IACOVONI J S,MARTINI H,et al. Kidney inflammaging is promoted by CCR2+ macrophages and tissue-derived micro-environmental factors[J]. Cell Mol Life Sci,2021,78(7):3485-3501. DOI:10.1007/s00018-020-03719-0.
FUJIU K,MANABE I,NAGAI R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice[J]. J Clin Investig,2011,121(9):3425-3441. DOI:10.1172/JCI57582.
HEARPS A C,MARTIN G E,ANGELOVICH T A,et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function[J]. Aging Cell,2012,
11(5):867-875. DOI:10.1111/j.1474-9726.2012.00851.x.
ZHANG B,BAILEY W M,BRAUN K J,et al. Age decreases macrophage IL-10 expression:implications for functional recovery and tissue repair in spinal cord injury[J]. Exp Neurol,2015,273:83-91. DOI:10.1016/j.expneurol.2015.08.001.
HOLT D J,GRAINGER D W. Senescence and quiescence induced compromised function in cultured macrophages[J]. Biomaterials,2012,33(30):7497-7507. DOI:10.1016/j.biomaterials.2012.06.099.
BOADA-ROMERO E,MARTINEZ J,HECKMANN B L,et al. The clearance of dead cells by efferocytosis[J]. Nat Rev Mol Cell Biol,2020,21(7):398-414. DOI:10.1038/s41580-020-0232-1.
DORAN A C,YURDAGUL A Jr,TABAS I. Efferocytosis in health and disease[J]. Nat Rev Immunol,2020,20(4):254-267. DOI:10.1038/s41577-019-0240-6.
MORIOKA S,MAUER?DER C,RAVICHANDRAN K S. Living on the edge:efferocytosis at the interface of homeostasis and pathology[J]. Immunity,2019,50(5):1149-1162. DOI:10.1016/j.immuni.2019.04.018.
SCHLOESSER D,LINDENTHAL L,SAUER J,et al. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis[J]. J Cell Biol,2023,222(2):e202207097. DOI:10.1083/jcb.202207097.
ORECCHIONI M,GHOSHEH Y,PRAMOD A B,et al. Macrophage polarization:different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages[J]. Front Immunol,2019,10:1084. DOI:10.3389/fimmu.2019.01084.
MONTANARO M,MELONI M,ANEMONA L,et al. Macrophage activation and M2 polarization in wound bed of diabetic patients treated by dermal/epidermal substitute nevelia[J]. Int J Low Extrem Wounds,2022,21(4):377-383. DOI:10.1177/1534734620945559.
TAMAKI S,KUROSHIMA S,HAYANO H,et al. Dynamic polarization shifting from M1 to M2 macrophages in reduced osteonecrosis of the jaw-like lesions by cessation of anti-RANKL antibody in mice[J]. Bone,2020,141:115560. DOI:10.1016/j.bone.2020.115560.
KOHNO K,KOYA-MIYATA S,HARASHIMA A,et al. Inflammatory M1-like macrophages polarized by NK-4 undergo enhanced phenotypic switching to an anti-inflammatory M2-like phenotype upon co-culture with apoptotic cells[J]. J Inflamm,2021,18(1):2. DOI:10.1186/s12950-020-00267-z.
DING L,YUAN X Y,YAN J H,et al. Nrf2 exerts mixed inflammation and glucose metabolism regulatory effects on murine RAW264.7 macrophages[J]. Int Immunopharmacol,2019,71:198-204. DOI:10.1016/j.intimp.2019.03.023.
SPRANGERS S,DE VRIES T J,EVERTS V. Monocyte heterogeneity:consequences for monocyte-derived immune cells[J]. J Immunol Res,2016,2016:1475435. DOI:10.1155/2016/1475435.
GEBRAAD A,KORNILOV R,KAUR S,et al. Monocyte-derived extracellular vesicles stimulate cytokinesecretion and gene expression of matrixmetalloproteinases by mesenchymal stem/stromal cells[J]. FEBS J,2018,285(12):2337-2359. DOI:10.1111/febs.14485.
ONG S M,HADADI E,DANG T M,et al. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence[J]. Cell Death Dis,2018,9(3):266. DOI:10.1038/s41419-018-0327-1.
PENCE B D,YARBRO J R,EMMONS R S. Growth differentiation factor-15 is associated with age-related monocyte dysfunction[J]. Aging Med,2021,4(1):47-52. DOI:10.1002/agm2.12128.
TSUKASAKI M,KOMATSU N,NAGASHIMA K,et al. Host defense against oral microbiota by bone-damaging T cells[J]. Nat Commun,2018,9(1):701. DOI:10.1038/s41467-018-03147-6.
GARLET G P,CARDOSO C R,MARIANO F S,et al. Regulatory T cells attenuate experimental periodontitis progression in mice[J]. J Clin Periodontol,2010,37(7):591-600. DOI:10.1111/j.1600-051X.2010.01586.x.
KOMATSU N,TAKAYANAGI H. Immune-bone interplay in the structural damage in rheumatoid arthritis[J]. Clin Exp Immunol,2018,194(1):1-8. DOI:10.1111/cei.13188.
JANG H M,PARK J Y,LEE Y J,et al. TLR2 and the NLRP3 inflammasome mediate IL-1β production in Prevotella nigrescens-infected dendritic cells[J]. Int J Med Sci,2021,18(2):432-440. DOI:10.7150/ijms.47197.
ELSAYED R,ELASHIRY M,LIU Y T,et al. Porphyromonas gingivalis provokes exosome secretion and paracrine immune senescence in bystander dendritic cells[J]. Front Cell Infect Microbiol,2021,11:669989. DOI:10.3389/fcimb.2021.669989.
S?DERSTR?M K,STEIN E,COLMENERO P,et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis[J]. Proc Natl Acad Sci U S A,2010,107(29):13028-13033. DOI:10.1073/pnas.1000546107.
ZANG J F,YE J,ZHANG C,et al. Senescent hepatocytes enhance natural killer cell activity via the CXCL-10/CXCR3 axis[J]. Exp Ther Med,2019,18(5):3845-3852. DOI:10.3892/etm.2019.8037.
RAJAGOPALAN S,LEE E C,DUPRIE M L,et al. TNFR-associated factor 6 and TGF-β-activated kinase 1 control signals for a senescence response by an endosomal NK cell receptor[J]. J Immunol,2014,192(2):714-721. DOI:10.4049/jimmunol.1302384.
RAJAGOPALAN S,LONG E O. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling[J]. Proc Natl Acad Sci U S A,2012,109(50):20596-20601. DOI:10.1073/pnas.1208248109.
DAR H Y,SINGH A,SHUKLA P,et al. High dietary salt intake correlates with modulated Th17-Treg cell balance resulting in enhanced bone loss and impaired bone-microarchitecture in male mice[J]. Sci Rep,2018,8(1):2503. DOI:10.1038/s41598-018-20896-y.
DAR H Y,SHUKLA P,MISHRA P K,et al. Lactobacillus acidophilus inhibits bone loss and increases bone heterogeneity in osteoporotic mice via modulating Treg-Th17 cell balance[J]. Bone Rep,2018,8:46-56. DOI:10.1016/j.bonr.2018.02.001.
SHASHKOVA E V,TRIVEDI J,CLINE-SMITH A B,et al. Osteoclast-primed Foxp3+ CD8 T cells induce T-bet,eomesodermin,and IFN-γ to regulate bone resorption[J]. J Immunol,2016,197(3):726-735. DOI:10.4049/jimmunol.1600253.
FUKUSHIMA Y,MINATO N,HATTORI M. The impact of senescence-associated T cells on immunosenescence and age-related disorders[J]. Inflamm Regen,2018,38:24. DOI:10.1186/s41232-018-0082-9.
CALLENDER L A,CARROLL E C,BEAL R W J,et al. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK[J]. Aging Cell,2018,
17(1):e12675. DOI:10.1111/acel.12675.
FRASCA D,DIAZ A,ROMERO M,et al. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways[J]. Exp Gerontol,2017,87(Pt A):113-120. DOI:10.1016/j.exger.2016.12.001.
LI Y,TERAUCHI M,VIKULINA T,et al. B cell production of both OPG and RANKL is significantly increased in aged mice[J]. Open Bone J,2014,6:8-17. DOI:10.2174/1876525401406010008.
ZHANG Z,YUAN W,DENG J J,et al. Granulocyte colony stimulating factor (G-CSF) regulates neutrophils infiltration and periodontal tissue destruction in an experimental periodontitis[J]. Mol Immunol,2020,117:110-121. DOI:10.1016/j.molimm.2019.11.003.
BREUIL V,TICCHIONI M,TESTA J,et al. Immune changes in post-menopausal osteoporosis:the Immunos study[J]. Osteoporos Int,2010,21(5):805-814. DOI:10.1007/s00198-009-1018-7.
BHAUMIK D,SCOTT G K,SCHOKRPUR S,et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8[J]. Aging,2009,1(4):402-411. DOI:10.18632/aging.100042.
LANG A,GRETHER-BECK S,SINGH M,et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4[J]. Aging,2016,8(3):484-505. DOI:10.18632/aging.100905.
NOH J H,KIM K M,IDDA M L,et al. GRSF1 suppresses cell senescence[J]. Aging,2018,10(8):1856-1866. DOI:10.18632/aging.101516.
NELSON G,KUCHERYAVENKO O,WORDSWORTH J,et al. The senescent bystander effect is caused by ROS-activated NF-κB signalling[J]. Mech Ageing Dev,2018,170:30-36. DOI:10.1016/j.mad.2017.08.005.
NACARELLI T,LAU L,FUKUMOTO T,et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome[J]. Nat Cell Biol,2019,21(3):397-407. DOI:10.1038/s41556-019-0287-4.
KIM S J,MEHTA H H,WAN J X,et al. Mitochondrial peptides modulate mitochondrial function during cellular senescence[J]. Aging,2018,10(6):1239-1256. DOI:10.18632/aging.101463.
PLAFKER K S,ZYLA K,BERRY W,et al. Loss of the ubiquitin conjugating enzyme UBE2E3 induces cellular senescence[J]. Redox Biol,2018,17:411-422. DOI:10.1016/j.redox.2018.05.008.
VIZIOLI M G,LIU T H,MILLER K N,et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence[J]. Genes Dev,2020,34(5/6):428-445. DOI:10.1101/gad.331272.119.
JOY J,BARRIO L,SANTOS-TAPIA C,et al. Proteostasis failure and mitochondrial dysfunction leads to aneuploidy-induced senescence[J]. Dev Cell,2021,56(14):2043-2058.e7. DOI:10.1016/j.devcel.2021.06.009.
YAMASHITA R,FUJII S,USHIODA R,et al. Ca2+ imbalance caused by ERdj5 deletion affects mitochondrial fragmentation[J]. Sci Rep,2021,11(1):20772. DOI:10.1038/s41598-021-99980-9.
GAN X Q,HUANG S B,YU Q,et al. Blockade of Drp1 rescues oxidative stress-induced osteoblast dysfunction[J]. Biochem Biophys Res Commun,2015,468(4):719-725. DOI:10.1016/j.bbrc.2015.11.022.
ZHANG L,GAN X Q,HE Y T,et al. Drp1-dependent mitochondrial fission mediates osteogenic dysfunction in inflammation through elevated production of reactive oxygen species[J]. PLoS One,2017,12(4):e0175262. DOI:10.1371/journal.pone.0175262.
JEONG S,SEONG J H,KANG J H,et al. Dynamin-related protein 1 positively regulates osteoclast differentiation and bone loss[J]. FEBS Lett,2021,595(1):58-67. DOI:10.1002/1873-3468.13963.
BADER V,WINKLHOFER K F. PINK1 and Parkin:team players in stress-induced mitophagy[J]. Biol Chem,2020,401(6/7):891-899. DOI:10.1515/hsz-2020-0135.
LEE S Y,AN H J,KIM J M,et al. PINK1 deficiency impairs osteoblast differentiation through aberrant mitochondrial homeostasis[J]. Stem Cell Res Ther,2021,12(1):589. DOI:10.1186/s13287-021-02656-4.
WANG X,LI H,ZHENG A,et al. Mitochondrial dysfunction-associated OPA1 cleavage contributes to muscle degeneration:preventative effect of hydroxytyrosol acetate[J]. Cell Death Dis,2014,5(11):e1521. DOI:10.1038/cddis.2014.473.
CAI W J,CHEN Y,SHI L X,et al. AKT-GSK3 β signaling pathway regulates mitochondrial dysfunction-associated OPA1 cleavage contributing to osteoblast apoptosis:preventative effects of hydroxytyrosol[J]. Oxid Med Cell Longev,2019,2019:4101738. DOI:10.1155/2019/4101738.
MAO Y X,CAI W J,SUN X Y,et al. RAGE-dependent mitochondria pathway:a novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products[J]. Cell Death Dis,2018,9(6):674. DOI:10.1038/s41419-018-0718-3.
WANG W D,KANG W B,ZHOU X Q,et al. Mitochondrial protein OPA mediates osteoporosis induced by radiation through the P38 signaling pathway[J]. Eur Rev Med Pharmacol Sci,2018,22(23):8091-8097. DOI:10.26355/eurrev_201812_16499.
MIDHA A,PAN H,ABARCA C,et al. Unique human and mouse β-cell senescence-associated secretory phenotype (SASP) reveal conserved signaling pathways and heterogeneous factors[J]. Diabetes,2021,70(5):1098-1116. DOI:10.2337/db20-0553.
BRAWERMAN G,NTRANOS V,THOMPSON P J. Alpha cell dysfunction in type 1 diabetes is independent of a senescence program[J]. Front Endocrinol,2022,13:932516. DOI:10.3389/fendo.2022.932516.
BAHOUR N,BLEICHMAR L,ABARCA C,et al. Clearance of p16Ink4a-positive cells in a mouse transgenic model does not change β-cell mass and has limited effects on their proliferative capacity[J]. Aging,2023,15(2):441-458. DOI:10.18632/aging.204483.
PRATTICHIZZO F,DE NIGRIS V,MANCUSO E,et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages[J]. Redox Biol,2018,15:170-181. DOI:10.1016/j.redox.2017.12.001.
WANG Q,NIE L,ZHAO P F,et al. Diabetes fuels periodontal lesions via GLUT1-driven macrophage inflammaging[J]. Int J Oral Sci,2021,13(1):11. DOI:10.1038/s41368-021-00116-6.
ZHANG P,WANG Q,NIE L,et al. Hyperglycemia-induced inflamm-aging accelerates gingival senescence via NLRC4 phosphorylation[J]. J Biol Chem,2019,294(49):18807-18819. DOI:10.1074/jbc.RA119.010648.
FRASCA D,ROMERO M,DIAZ A,et al. B cells with a senescent-associated secretory phenotype accumulate in the adipose tissue of individuals with obesity[J]. Int J Mol Sci,2021,22(4):1839. DOI:10.3390/ijms22041839.
RABHI N,DESEVIN K,BELKINA A C,et al. Obesity-induced senescent macrophages activate a fibrotic transcriptional program in adipocyte progenitors[J]. Life Sci Alliance,2022,5(5):e202101286. DOI:10.26508/lsa.202101286.
BOULET N,BRIOT A,JARGAUD V,et al. Notch activation shifts the fate decision of senescent progenitors toward myofibrogenesis in human adipose tissue[J]. Aging Cell,2023,22(3):e13776. DOI:10.1111/acel.13776.
XU M,TCHKONIA T,DING H S,et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age[J]. Proc Natl Acad Sci U S A,2015,112(46):E6301-E6310. DOI:10.1073/pnas.1515386112.
MANDL M,WAGNER S A,HATZMANN F M,et al. Sprouty1 prevents cellular senescence maintaining proliferation and differentiation capacity of human adipose stem/progenitor cells[J]. J Gerontol A Biol Sci Med Sci,2020,75(12):2308-2319. DOI:10.1093/gerona/glaa098.
CONLEY S M,HICKSON L J,KELLOGG T A,et al. Human obesity induces dysfunction and early senescence in adipose tissue-derived mesenchymal stromal/stem cells[J]. Front Cell Dev Biol,2020,8:197. DOI:10.3389/fcell.2020.00197.
RATUSHNYY A,EZDAKOVA M,BURAVKOVA L. Secretome of senescent adipose-derived mesenchymal stem cells negatively regulates angiogenesis[J]. Int J Mol Sci,2020,21(5):1802. DOI:10.3390/ijms21051802.
LI Y J,LU L Y,XIE Y,et al. Interleukin-6 knockout inhibits senescence of bone mesenchymal stem cells in high-fat diet-induced bone loss[J]. Front Endocrinol,2020,11:622950. DOI:10.3389/fendo.2020.622950.
VALVERDE M,S?NCHEZ-BRITO A. Sustained activation of TNFα-induced DNA damage response in newly differentiated adipocytes[J]. Int J Mol Sci,2021,22(19):10548. DOI:10.3390/ijms221910548.
VELDHUIS-VLUG A G,ROSEN C J. Clinical implications of bone marrow adiposity[J]. J Intern Med,2018,283(2):121-139. DOI:10.1111/joim.12718.
TENCEROVA M,FIGEAC F,DITZEL N,et al. High-fat diet-induced obesity promotes expansion of bone marrow adipose tissue and impairs skeletal stem cell functions in mice[J]. J Bone Miner Res,2018,33(6):1154-1165. DOI:10.1002/jbmr.3408.
LIU X N,GU Y R,KUMAR S,et al. Oxylipin-PPARγ-initiated adipocyte senescence propagates secondary senescence in the bone marrow[J]. Cell Metab,2023,35(4):667-684.e6. DOI:10.1016/j.cmet.2023.03.005.
(收稿日期:2023-09-25;修回日期:2024-01-20)
(本文编辑:赵跃翠)
猜你喜欢
中成药(2017年5期)2017-06-13 13:01:12
上海农业学报(2017年4期)2017-04-10 12:40:22
中国实用医药(2016年27期)2016-11-30 11:17:40
中国民族民间医药·上半月(2016年10期)2016-11-19 11:22:25
科技资讯(2016年19期)2016-11-15 10:37:59
上海医药(2016年17期)2016-10-12 01:55:41
天然产物研究与开发(2016年6期)2016-06-05 10:29:30
中成药(2016年8期)2016-05-17 06:08:15
养生保健指南(2016年4期)2016-03-22 12:39:35
养生保健指南(2016年4期)2016-03-22 12:25:57
