
高中生物学PCR技术中的引物设计
2024-07-08 17:18陈铁鑫
中学教学参考·理科版 2024年5期
关键词:高中生物学
陈铁鑫


[摘 要]聚合酶链式反应(PCR)是江苏高考生物试题中的常考内容,PCR技术的用途并不局限于最初、最基本的获取目的基因。PCR技术的使用场景极其灵活,通过引物设计,可以达到融合基因、定点诱变等效果。文章对PCR技术中的引物设计思路进行分类,介绍其衍生的各种应用,为教师教学提供参考。
[关键词]PCR;引物设计;高中生物学
[中图分类号] G633.91 [文献标识码] A [文章编号] 1674-6058(2024)14-0084-04
核心素养是高考命题和各类考试命题的指导方向和原则。为加深学生对基因工程大概念的理解,提升学生的生物学科核心素养,《普通高中生物学课程标准(2017年版2020年修订)》要求教师在基因工程的教学过程中开展“利用聚合酶链式反应(PCR)扩增DNA片段并完成电泳鉴定,或运用软件进行虚拟PCR实验”活动[1]。引物直接决定PCR的特异性与成功与否。人教版高中生物学选择性必修三中通过图片简单演示了PCR的基本过程,并未涉及引物设计,而近几年各地的高考试题为了响应新课标要求,基于核酸检测、DNA测序等真实情境,增加了许多与引物设计相关的技术困境内容。为跨越教材与实际应用的鸿沟,笔者对不同应用场景中的引物设计进行分析,以促进学生对PCR的理解,为教师教学提供参考。
一、以基因定位为目的的反向引物设计
以真核生物为受体细胞时,目的基因一般需整合至受体细胞的染色体DNA中,以实现稳定存在与表达。在上述情境中,为进行目的基因定位,需要在已知目的基因的附近逐步搜索并测序相邻的DNA区域,这就需要用到反向PCR。
[例1]农杆菌Ti质粒上的T-DNA可以转移并随机插入到被侵染植物的DNA中。研究者将图1中被侵染植物的DNA片段连接成环,并以此环为模板进行PCR,扩增出T-DNA插入位置两侧的未知序列,以此可确定T-DNA插入的具体位置。下列说法不正确的是()。
图 1
A.PCR扩增依据的是DNA分子双链复制的原理
B.进行PCR扩增需要热稳定DNA聚合酶
C.利用图中的引物②、③组合可扩增出两侧的未知序列
D.通过与受体细胞的基因组序列比对,可确定T-DNA的插入位置
解析:对含目的基因的T-DNA中两侧未知序列通过同种限制酶或是同尾酶进行酶切(目的基因中不能存在该酶的酶切位点),可取得相同的黏性末端。通过DNA连接酶令图 1含T-DNA的片段自身环化[2]成图2甲的环形DNA分子。
碍于DNA聚合酶的特性,子链总是从5'端往3'端延伸。若目的为扩增T-DNA,应选择指向已知扩增片段的内侧的引物②、③。选择引物①、④,子链向外侧未知序列进行延伸。由于PCR体系中没有DNA连接酶,因此子代DNA仍是线性(如图2乙)。扩增后,原本在两侧的未知序列被转移到已知序列的内部(如图2丙)。随后,我们可以对未知序列进行测序,比对受体细胞的基因组文库,即可确定T-DNA插入的位置。
图 2
参考答案:C
二、鉴定目的基因连接方式的反向引物设计
构建基因表达载体时,若使用单酶切可能出现目的基因的反向连接。目的基因的筛选与检测是基因工程的重要环节,可以通过设计引物进行PCR,筛选目的基因与载体的正向连接产物。
[例2]为鉴定筛选出的菌落中是否含有正确插入目的基因的重组质粒,拟设计引物进行PCR鉴定。图3所示为甲、乙、丙3条引物在正确重组质粒中的相应位置。
(1)PCR鉴定时应选择的一对引物是 。
(2)某学生尝试用图中另外一对引物从某一菌落的质粒中扩增出了400 bp片段,原因是 。
解析:如图 4所示,黑色区域为目的基因,我们在目的基因内、外各设计一引物,图中为正向连接时各引物序列的位置。使用引物1、引物3可扩增出对应片段,当目的基因反向连接时,引物1与引物3方向相同,无法有效扩增。同理,选择引物2、引物4,也可以进行上述验证。与预期相符的DNA长度就可以用于确认目的基因的正向连接。
图3为正向连接时的引物分布。引物丙、引物甲位于目的基因的上游和下游,无论连接方向是否正确,均可扩增出片段且长度相同,无法作为排除的依据。
若使用引物丙、引物乙进行PCR,考虑反向连接时,引物乙的方向与引物丙相同,无法扩增片段,正向连接时引物丙、引物乙可以扩增出350 bp长度的片段。
同理,若选择引物甲、引物乙进行PCR,反向连接时能扩增出400 bp片段,正向连接时无扩增产物,但空质粒也会带来相同的结果,无法确定实现了正向连接。
因此,PCR鉴定时应选择引物乙、引物丙。学生扩增出400 bp片段的原因是目的基因反向连接。
参考答案:(1)乙、丙 (2)目的基因反向连接
三、PCR体系中的引物序列改造
目的基因想要导入表达载体,两侧需要有合适的酶切位点。实验室用到的限制酶价格高昂,种类有限,为了不被基因自身序列限制,就需要对引物序列进行改造。引物设计的依据是目的基因的两侧序列,但这并不意味着引物需要与模板完全互补。我们既可以在5'端进行添加限制酶识别序列,也可以在中间进行少量碱基的增删或者替换。引物改造的部分序列不能和原对应模板配对,但不影响引物和模板的整体配对,因此,其扩增产物的对应位置就会引入识别序列,或是发生碱基对的增删或者替换[3]。
(一)引入序列与连接DNA
引物与模板的结合并不需要完全一一对应。引物的3'端是DNA聚合酶添加游离脱氧核苷酸的位点,如果引入的核苷酸序列添加在3'端,会导致扩增产物的特异性下降。因此,我们常常只在5'端添加所需要的酶切位点。若在两个DNA片段对应引物的5'端分别构建相同或者互补序列,对获得的新DNA进行变性与杂交,还可以达到连接DNA的效果。
[例3]科研人员通过敲除里氏木霉基因组中去乙酰化酶基因(HDAC),探究组蛋白乙酰化对纤维素酶基因表达的影响,其主要过程如图5。请据图回答:引物设计是PCR的关键,本研究中引物R1的5'端添加与引物FG互补的序列,其目的是 。在PCR5中加入的引物是 。
图 5
解析:通过PCR1、PCR2,可获得新霉素抗性基因GR与H1的构造(如图 6)。
图 6
以GR 基因、H1为模板,F1和RG为引物进行PCR4扩增,在高温作用下双链会打开。由题可知,引物R1的5'端添加了与引物FG互补的序列,因此其延伸产物H1基因的b链5'端也存在引物FG的互补序列,b链与c链就能够部分配对(如图 7)。Taq DNA聚合酶只能从5'端向3'端延伸,这两条链的3'端都缺乏模板,无法继续延伸。
图 7
a链的3'端与R1序列互补,d链的3'端与FG互补。由于R1与FG互补,因此a链的3'端和d链的3'端也是互补的(如图 8)。
图 8
在Taq DNA聚合酶的作用下,可将重叠的两条链补齐(如图9)。
图 9
两个基因就这样融合成功。随后a链、d链分别结合引物RG、F1进行PCR4,就得到了大量的H1-GR融合基因。这种借助引入序列连接DNA的方法我们称为重叠延伸PCR。按照相同的逻辑,可以将基因H2和GR也进行连接。
图10
如图10,通过类比,不难想到应令c链和f链发生互补配对、延伸。随后c链、f链分别以R2、FG为对应引物,进行大量扩增。为确保这两条链的碱基互补配对,可在F2的5'端通过引入序列,添加与RG互补的序列。
综上,本研究中在引物R1的5'端添加与引物FG互补的序列,其目的是便于PCR4时H1和GR基因的连接。在PCR5中加入的引物是R2和FG。
参考答案:便于H1和GR的拼接 R2和FG
(二)定点诱变
引物是PCR产物5'端的一部分,引物长度一般为15~27 bp,因此最初建立的PCR定点诱变技术只适用于DNA的末端突变。突变位点在基因中部时,我们就得设计两对引物,基因中部的两个诱变引物分别与两侧常规引物将目的基因分段扩增,再以上文中连接DNA的方式将两段诱变DNA融合。
[例4]重叠延伸PCR技术是采用具有互补末端的引物,使PCR产物形成重叠链,从而在随后的扩增反应中通过重叠链的延伸,获得想要的目的基因。某科研团队运用重叠延伸PCR技术在水蛭素基因的特定位点引入特定突变,以实现基因的定点突变,原理如图11。下列说法错误的是()。
图11
A. 过程②需要含Mg2+的缓冲液、DNA模板、引物、四种脱氧核苷酸、耐高温的DNA聚合酶等
B. 若引物1、引物2组成的反应系统和引物3、引物4组成的反应系统中均进行一次DNA分子的复制后,一共会产生3种DNA分子
C. 经过过程④获得的杂交DNA有2种,其中只有一种可以经过过程⑤获得目的基因
D. 过程⑤使用耐高温的DNA聚合酶延伸,需要引物
解析:过程④获得的杂交DNA具有游离的3'端,也具有模板,可以直接使用耐高温的DNA聚合酶延伸,不需要引物。其连接机制与例3相同。
参考答案:D
四、特异性扩增的嵌套引物设计
引物的长度一般在20个碱基左右,因此从概率上讲,待扩增片段上出现与引物配对序列的概率是[1420],约万亿分之一,因此一段长度为20 bp的序列在人类基因组约31.6亿碱基对的排列组合中理论上重复的概率很低。但碱基对的排列顺序并不是完全随机的,引物与模板链的结合也并不需要完全互补,因此总有非特异性扩增条带的出现。巢式PCR为了解决上述困境,对PCR体系中的引物数量进行了设计。
[例5]普通PCR在扩增DNA的过程中存在一定概率的碱基错误配对,得到部分的错误产物。巢式PCR是一种在普通PCR基础上改良的新型PCR技术,扩增的精确性比普通PCR更高。巢式PCR对目标DNA进行扩增时需要用到两对不同的引物,首先使用第一对引物(外引物)对目标DNA进行第一次扩增得到中间产物,然后使用第二对引物(内引物)对中间产物进行第二次扩增得到目标产物,过程如图12所示。请回答相关问题:
图12
巢式PCR通常在第一支试管中进行第一次扩增,然后把得到的中间产物分离出来转移到另外一支试管中进行第二次扩增反应,两次扩增不在同一支试管中扩增的原因是 。
解析:针对已知的目的基因两侧部分序列,设计外引物1、4,内引物2、3。
图13
如图13,首先通过外引物1、4扩增序列,此时依然存在这样的可能性:引物与其他未知片段结合。经过第一次扩增后,提取反应产物作为下一轮模板,使用内引物2、3进行第二次扩增。若第一次扩增出非特异性条带,第二次扩增因缺乏模板无法正常进行。第二次扩增的成功也是对第一次扩增反应正常进行的鉴定。因此,若两次扩增在同一个反应体系中进行,精确性就无法得到验证。
参考答案:防止内引物直接参与第一次扩增,无法实现巢式PCR高精确性扩增
五、利用浓度不对称的引物制备DNA单链
基因组DNA分析经常用到southern印迹,依赖单链DNA探针与固定的DNA杂交。为了制备扩增特异长度的单链DNA,我们采用不同浓度的引物。高浓度引物的量是低浓度引物的50~100倍。在一个PCR体系中,低浓度引物被快速消耗完后,高浓度引物主导之后的PCR进程,从而产生大量单链DNA。
[例6]DNA分子杂交时,常需要大量的单链DNA探针。不对称PCR是一种常见的单链DNA探针制备方法,其原理是反应体系中加入一对数量不相等的引物。在PCR反应的早期10~15个循环中,扩增产物主要是双链DNA,当后期数量较少的限制性引物耗尽后,数量较多的非限制性引物将引导合成大量目标DNA单链。
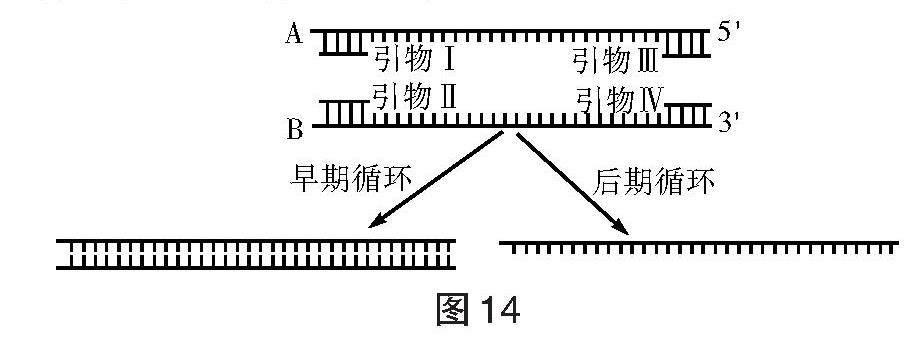
图14
若图14中A链为所需的DNA分子探针,则非限制性引物应选用引物 。
解析:若选择引物Ⅱ、Ⅲ,限于子链延伸方向向外,扩增的是无效片段。引物Ⅳ与B链互补,A链与B链互补,故引物Ⅳ是合成大量A链所必不可少的一部分。为合成大量A链,就需要大量引物Ⅳ作为非限制性引物。通过最初10~15个循环,获得大量模板B链 。后15次循环只以大量B链为模板产生大量A链作探针。单链的分子量较小,通过电泳可以将这些单链DNA探针分离出来。
参考答案:Ⅳ
六、检测特定基因的存在或缺失的多重PCR
一个抗原可能具备多个抗原决定簇,对应多个基因位点。有时需要检测一份样本中是否含有多种病原体,或者对具有多个位点的病毒进行探究,如SARS-CoV-2的检测诊断至少需要两个基因靶标[4]。这种情况下,为节约人力物力,并且高效快速检测,可以在同一反应体系中加入两对以上的引物。这种方式称为多重PCR,其优点是可以通过一次PCR同时对多个目的序列进行扩增,还可以检测特定基因序列的存在或缺失。
[例7]人类γ基因启动子上游的调控序列中含有BCL11A蛋白结合位点,该位点结合BCL11A蛋白后,γ基因的表达被抑制。通过改变该结合位点的序列,解除对γ基因表达的抑制,可对某种地中海贫血症进行基因治疗。科研人员扩增了γ基因上游不同长度的片段,将这些片段分别插入表达载体中进行转化和荧光检测,以确定BCL11A蛋白结合位点的具体位置。F1~F7,R均为引物。相关信息如图15所示。
图15
含F1~F4与R扩增产物的受体细胞不再有荧光,而含F5~F7与R扩增产物的受体细胞仍有荧光。若γ基因上游调控序列上与引物序列所对应的位置不含有BCL11A蛋白的结合位点序列,据此结果可推测,BCL11A蛋白结合位点位于 。
解析:荧光蛋白终止子在DNA左侧,为使荧光蛋白基因表达,应将调控序列及启动子反向连接在荧光蛋白基因与启动子P之间。参考图16。
图16
若扩增产物含有BCL11A蛋白结合位点,该位点与BCL11A蛋白的结合将抑制荧光蛋白的表达,进而使得受体细胞不能发出荧光。使用F5、R扩增的片段仍有荧光,因此无结合位点。使用F4、R扩增的片段荧光消失,因此结合位点在F4~F5对应序列之间的区段。
参考答案:F4~F5对应序列之间的区段
[ 参 考 文 献 ]
[1] 中华人民共和国教育部.普通高中生物学课程标准:2017年版2020年修订[M].北京:人民教育出版社,2020.
[2] 屈伸,刘志国.分子生物学实验技术[M].北京:化学工业出版社,2008.
[3] 任桂杰,王志玉.PCR介导的定点突变与随机突变的应用[J].山东大学学报(医学版),2005(9):865-867.
[4] 李庆超,房学迅,徐小洁.多重PCR检测新型冠状病毒核酸的研究进展[J].军事医学,2022(10):798-801.
(责任编辑 罗 艳)
猜你喜欢
青年时代(2016年30期)2017-01-20
数学学习与研究(2016年19期)2016-11-22
中学生物学(2016年10期)2016-11-19
考试周刊(2016年73期)2016-09-21
求知导刊(2016年1期)2016-02-18
新课程·中旬(2015年10期)2015-11-30
