
猕猴桃溃疡病病原菌MLVA分型引物的筛选及验证
2024-06-30 12:38:12姚令王海黄露安星宇陈文王莉爽薛原吴石平
果树学报 2024年6期
姚令 王海 黄露 安星宇 陈文 王莉爽 薛原 吴石平

摘 要:【目的】筛选出一组可精准快速地对丁香假单胞菌猕猴桃致病变种(Pseudomonas syringae pv. actinidiae,Psa)进行分型的引物组合。【方法】针对前期文献已报道的34对引物,采用PCR技术验证该34对引物对中国Psa菌株的扩增效率及准确性;利用模拟PCR获取菌株串联重复(TR)数;以辛普森指数(Simpsons index,SI)作为筛选引物组合的标准,基于R软件平台,筛选最优引物组合。【结果】34对引物对中国Psa扩增效果均良好,其中TR14与TR11II、TR19与Psa-01引物序列相同;TR8与Psa-08、TR39II与Psa-10、GM-1834与TR10I、GM-1553与TR64II、TR19Psa-01与TR19II扩增同一TR;Psa-09扩增产物串联重复单元长度不唯一,TR2II扩增产物侧翼变异较大,不能通过电泳确定串联重复数;最终确定SI值与全部引物组合相同的最低引物数量为9对,使用该9对引物的组合可将Psa已知的5种生物型准确分开。【结论】TR23/Psa-04、Psa-03、Psa-05、Psa-06、TR10IGM-1834、TR30I、TR1II、Psa-10TR39II、TR64IIGM-1553等9对引物可代表当前文献报道的34对引物,进行Psa分型研究,探索猕猴桃溃疡病的传播和流行规律,为病害防控策略的制定提供科学依据。
关键词:丁香假单胞菌猕猴桃致病变种;多位点串联重复序列分析;群体遗传结构;引物;筛选
中图分类号:S663.4;S436.634 文献标志码:A 文章编号:1009-9980(2024)06-1188-11
Screening and validation of MLVA typing primers for the Pseudomonas syringae pv. actinidiae
YAO Ling1, WANG Hai2, HUANG Lu1, AN Xingyu1, CHEN Wen1, WANG Lishuang1, XUE Yuan3, WU Shiping1*
(1Institute of Plant Protection, Guizhou Academy of Agricultural Sciences/Key Laboratory of Crop Genetic Resources and Germplasm Innovation in Karst Mountains Ministry of Agriculture and Rural Affairs, Guiyang 550006, Guizhou, China; 2Institute of Agricultural Science and Technology Information, Guizhou Academy of Agricultural Sciences, Guiyang 550006, Guizhou, China; 3Anshun Branch of Guizhou Tobacco Company, Anshun 561000, Guizhou, China)
Abstract: 【Objective】 Kiwifruit canker caused by Pseudomonas syringae pv. actinidiae (Psa), is one of the most threatening diseases in the kiwifruit industry. Studying the population genetic structure of Psa can provide theoretical reference for scientific prevention and control of this disease. The multiple-locus variable-number tandem-repeats analysis (MLVA) has been reported to study the population genetic structure of Psa. At present, there are problems with inconsistent and excessive primers in the analysis of the genetic structure of Psa population using MLVA technology. Using too many primers will make MLVA typing technology lose its advantages like convenience and low cost. In order to screen primer combinations suitable for studying the population genetic structure of Psa, 34 pairs of primers reported were analyzed. 【Methods】 To verify the amplification efficiency of primers on Chinese Psa, we used 34 pairs of primers to amplify 10 strains of Psa isolated and preserved in our laboratory. We downloaded the whole genome data of 127 Psa strains from Genbank for primer screening. We then used 34 pairs of primers on the genome sequences of these 127 Psa strains to perform Simulated PCR to obtain TR data. Calculate MLGs and SI using “popper” package to evaluate the typing ability of each primer with Simpson index (SI) as the standard for screening primer combinations, develop a program code using R to calculate the SI of typing results for different primer combinations, and use the df2genind() function of the “poppr” package to convert the simulated PCR result data into genind format. Primer combination SI calculation was performed by using the diversity_stats [mlg.table()] function, starting from the SI of the typing results of 2 pairs of primer combinations, and then calculating 3 pairs of primer combinations until the SI of the calculated primer combination was equal to the SI of all primers. This primer combination was the optimal primer combination. Use the genotype_curve() function of the “poppr” package to statistically analyze the multi locus genotypes (MLGs) of Psa for all primer combinations; and using the entire genome sequences of 20 and 10 Psa strains, a UPGMA clustering tree was constructed using the bruvo.boot() function based on the selected primer combinations to verify the typing effect of the selected primer combinations. 【Results】 34 pairs of primers had good amplification efficiency for Psa in China. By analyzing the results of simulated PCR data, it was found that TR14 and TR11II, TR19 and Psa-01 had the same sequence. TR8 and Psa-08, TR39II and Psa-10, GM-1834 and TR10I, GM-1553 and TR64II, TR19Psa-01 and TR19II amplified the same TR. The TR unit length of Psa-09 amplification product was not unique and the lateral variation of TR2II amplification product was large, which was not determining TRs by electrophoresis. By calculating the MLGs of each primer typing result, it was found that 34 pairs of primer MLGs were between 2-20. Among them, TR10Ⅰ, GM-1834, TR39Ⅱ, and Psa-10 had the highest MLGs of 20, while TR15I, TR17 and TR22 had the lowest MLGs of 2; By calculating the SI of each primer typing result, it was found that the SI values of 34 pairs of primers ranged from 0.015 4 to 0.896 8, with TR10Ⅰ, GM-1834 having the highest SI value of 0.896 8 and TR15I having the lowest SI value of 0.015 4. Through the program developed by R, it was found that the SI value of different primer combination typing results using all primer combinations was 0.984 7. The combination of two pairs of primers with the highest SI was Psa03 and GM-1834TR10I, with an SI value of 0.973 6, which did not reach the SI value of all primers. The combination of three pairs of primers with the highest SI was Psa03, GM-1834TR10I, and TR39IIPsa10, with an SI value of 0.980 3, which still did not reach the SI value of all primers. The primer combinations with the same SI values as all primers were TR23/Psa-04, Psa-03, Psa-05, Psa-06, TR10IGM-1834, TR3II, TR1II, Psa-10TR39II and TR64IIGM-1553, among which TR23 and Psa04 can be replaced with each other, and you can choose one of them. The MLGs of the 9 primers typing results were equal to the MLGs of all primer typing results, UPGMA cluster tree analysis found that 9 pairs of primer combinations can accurately separate the 5 biovar of Psa, and some differences within the biovar 3 can be seen. 【Conclusion】 The above results indicated that using a combination of these 9 primers for typing analysis had the same effect as using all primers. After verification, the combination of 9 pairs of primers can accurately separate the 5 biovars of Psa. The combination of 9 pairs of primers including TR23/Psa-04, Psa-03, Psa-05, Psa-06, TR10IGM-1834, TR3II, TR1II, Psa-10TR39II and TR64IIGM-1553, can be used to study the population genetic structure of Psa, explore the transmission and prevalence patterns of kiwifruit canker disease, and provide scientific basis for the formulation of disease prevention and control strategies.
Key words: Pseudomonas syringae pv. actinidiae; Multiple loci variable number of tandem repeats analysis; Population genetic structure; Primer; Screen
丁香假单胞菌猕猴桃致病变种(Pseudomonas syringae pv. actinidiae,Psa)引起的猕猴桃细菌性溃疡病严重影响猕猴桃产业发展[1],该病害发生前期隐蔽性强,不易发现,且传播迅速、感染植株死亡率高,防治极其困难[2-3]。全国各猕猴桃产区均受到猕猴桃溃疡病的危害,受害严重园区发病率超过80%,甚至全园发病导致毁园[4-6]。近年来,各国研究者通过指纹图谱、多位点序列分析和全基因组序列分析等方法将Psa分为5种不同的生物型(biovar),分别为biovar 1、2、3、5和6。其中biovar 1最早在日本发现,意大利也曾有零星发现;biovar 5和6仅在日本发现;biovar 2仅在韩国发现;biovar 3为全球流行群体,其群体遗传结构十分复杂[7],中国截至目前所分离到的Psa均属于biovar 3。对Psa群体结构和分布特征开展系统分析有助于认识猕猴桃溃疡病的传播和流行趋势,为病害防控策略的制定提供科学依据[8-10]。
多位点可变数目串联重复序列分析(multiple loci variable number of tandem repeats analysis,MLVA)广泛应用于医学病原细菌及植物病原细菌群体遗传结构研究,具有成本低、分辨率高、数据易于整合、通量高等优点[11],Osanloo等[12]采用ERIC-PCR分型方法和MLVA分型方法对鲍曼不动杆菌开展分型研究,使用rep-PCR分型方法将伊朗100株鲍曼不动杆菌分为4个类群,MLVA分型方法则分为9个类群,表明MLVA技术分辨率较ERIC-PCR更高;Siarkou等[13]采用MLST分型和MLVA分型方法对流产衣原体进行了分型研究,发现MLST将流产衣原体分为6个类群,MLVA技术则分为7个类群,表明MLVA分型技术的分辨率高于MLST。已有研究者采用此方法对Psa群体遗传结构进行了研究,Ciarroni等[14]设计了13对MLVA引物,对142株Psa进行群体遗传结构研究,发现来自中国、日本、新西兰和法国等不同国家或地区的142株Psa可分为10个不同的亚群,并发现中国的Psa菌株具有广泛的遗传多样性;Cunty等[15]设计了11对MLVA引物对340株Psa的群体遗传结构进行了分析,发现中国Psa具有丰富的遗传多样性;赵志博等[16]设计了10对MLVA引物用以建立中国Psa群体遗传结构分型技术,并应用于来自贵州修文县7个代表性果园62株Psa群体结构研究中,发现62株Psa可分为3个不同的亚群。至此,MLVA引物数量多达34对。使用过多MLVA引物会使该技术失去低成本和便捷的优势。笔者在本研究中对已报道的MLVA引物进行筛选和分析,力求减少引物的使用而不降低分型效果,筛选出可以精准快速地进行Psa分型的引物组合。该引物组合可用于Psa群体遗传结构研究,探索Psa的传播和流行规律,为猕猴桃溃疡病防控策略的制定提供科学依据。
1 材料和方法
1.1 MLVA引物PCR扩增效率验证
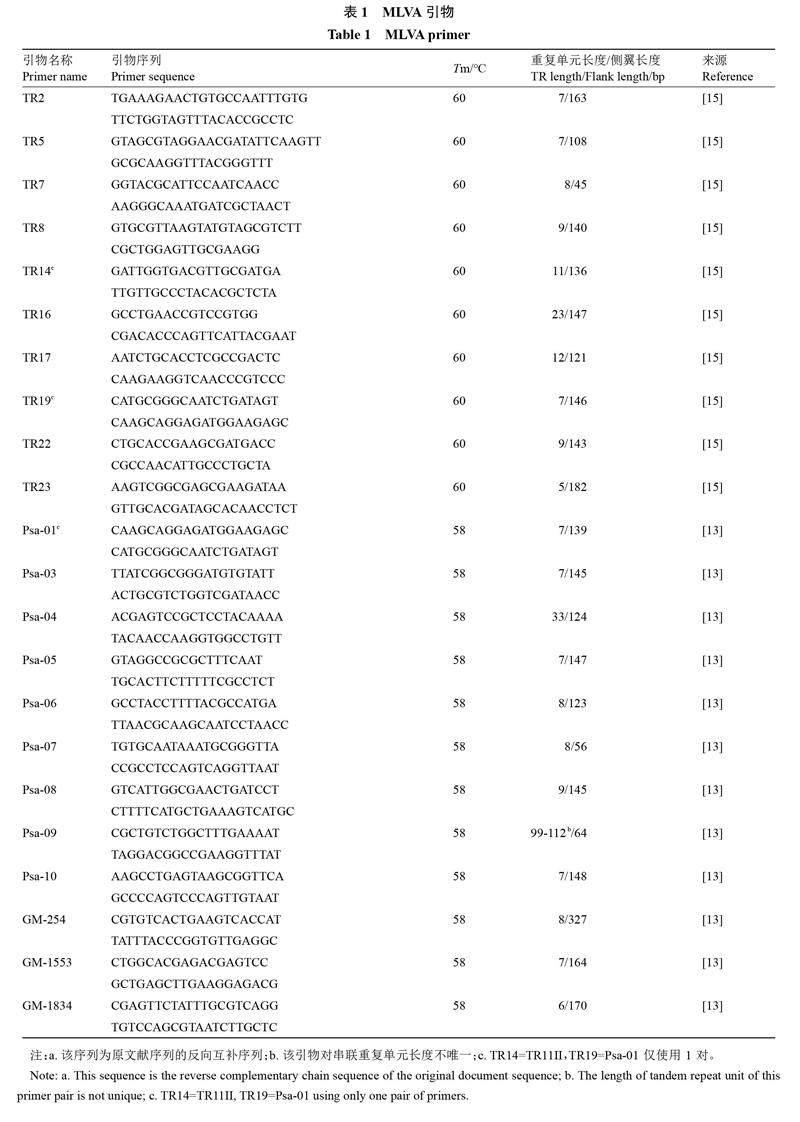
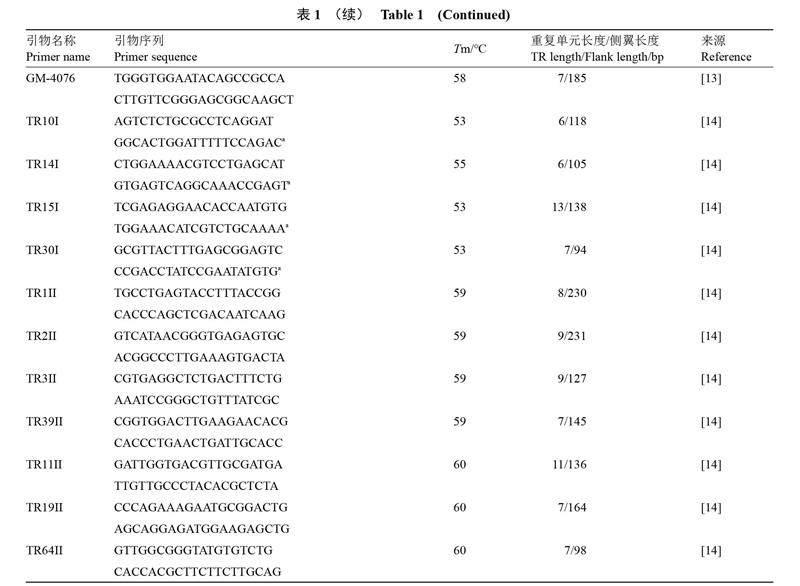
1.1.1 MLVA引物 MLVA引物如表1所示。
1.1.2 供试菌株 挑选10株具有代表性的Psa菌株验证34对引物对中国菌株的扩增效率(表2)。
1.1.3 PCR、电泳条件 PCR条件:95 ℃ 3 min,95 ℃ 30 s,退火温度30 s,72 ℃ 1.5 min,35个循环,72 ℃ 5 min,最后保持12 ℃(表1);毛细管电泳采用安捷伦科技有限公司生产的毛细管电泳仪,试剂采用DNF900试剂盒;毛细管电泳程序设置:5.0 kV 30 s,5.0 kV 10 s注入Marker,5.0 kV 10 s注入样品及Ladder,最后5.0 kV运行80 min。
1.2 模拟PCR筛选MLVA引物
以Genbank中下载127株Psa全基因组序列为模板,表1所列引物对为引物,使用SPCR 3.0进行模拟PCR,模拟PCR参数为:Up Threshold:0.8;Down Threshold:0.8;Pa Threshold:0.8;Max Product:500 bp;Min Product:35 bp(其中Psa-09 Max Product设置为1000 bp)[26]。
1.3 数据分析
1.3.1 串联重复数(TRs)计算 将模拟PCR扩增结果的长度信息导入Excel中,整理后按照下列公式进行串联重复数(tandem repeats,TRs)的计算,统计引物的扩增情况。
[串联重复数=序列长度-侧翼长度重复单元长度]。
1.3.2 引物组合筛选 以辛普森指数(Simpsons index,SI)作为筛选引物组合的标准,计算所有引物组合的SI,并根据SI的大小筛选引物组合[27]。基于R version 4.0.2开发程序进行引物组合的筛选,采用poppr软件包的df2genind()函数将模拟PCR结果矩阵转化为genind格式,利用diversity_stats [mlg.table()]函数进行引物组合SI的计算,从2对引物组合的SI开始计算,然后计算3对引物组合的SI,直到所计算引物组合的SI等于全部引物组合下的SI,该引物组合即为最优引物组合[28]。
1.3.3 引物组合效果验证 采用poppr软件包的genotype_curve()函数对所有的引物组合下Psa的多位点基因型数(Multilocus genotypes,MLGs)进行统计;以10株Psa的TR数据结合Genbank下载20株Psa全基因组序列(表2),采用bruvo.boot()函数基于筛选获得的引物组合构建UPGMA聚类树,验证所筛选引物组合的分型效果[29]。
2 结果与分析
2.1 PCR扩增效果验证
除TR10I、TR14I、TR15I、TR30I等4对引物外,Psa-10(见图1左)、TR2、TR5等30对引物对供试的10个Psa菌株均能扩增出清晰明亮的单一条带,进一步分析发现,TR10I(见图1右)、TR14I、TR15I、TR30I等4对引物的下游引物采用反向互补序列后可扩增出单一清晰条带。结果表明34对引物均对供试Psa菌株表现良好扩增效果,详细结果见表3。
2.2 模拟PCR
模拟PCR结果如表3所示,TR2等10对引物能将127株Psa完全扩增成功,GM-1834等22对引物扩增的菌株数大于105株,Psa-09和TR23引物扩增的菌株数最少,分别为59株和98株。
通过对模拟PCR扩增的序列文件分析,TR8与Psa-08、TR39II与Psa-10、GM-1834与TR10I、GM1553与TR64II、TR19Psa-01与TR19II扩增同一TR;Psa-09 TR长度不固定,TR2II侧翼长度变异较大,不能通过电泳准确推断TR数。将序列相同的引物、扩增同一TR的引物仅保留1对,不能通过电泳判断TRs的引物被剔除,剩余25对引物进行后续的分析。
25对引物扩增的产物长度、多位点基因型数(MLGs)、辛普森指数(SI)结果各不相同。从MLGs来看TR10I、GM-1834、Psa-10和TR39II等4对引物扩增的MLGs最多,为20个;TR15I、TR17和TR22等3对引物扩增的MLGs最少,为2个;从SI来看,TR39IIPsa-10、TR10I、GM-1834和Psa-03等4对引物扩增基因型的SI较高,分别为0.896 8、0.878 9、0.878 9和0.735 3;TR15I、TR30I、GM-4076和TR3II等4对引物扩增基因型的SI较低,均小于0.3。25对引物中,GM-254扩增的MLGs较多,为9,其SI为0.356 2,均匀度较低;TR17和TR22扩增的MLGs较少,为2,SI为0.478 1,均匀度较高(表4)。
2.3 引物组合筛选
使用R进行25对引物所有组合的查找及SI值的计算结果表明:选择25对引物时,其组合仅有1个,SI值为0.984 7;选择9对引物时,引物组合有2 042 975个,其SI值最大的前3个组合的SI值分别为0.984 7、0.984 7和0.984 5,最大SI值与25对引物的SI值相同。选择9对以上引物组合时,其最大SI值也达到了0.984 7,最大SI值未随引物增加而增加(表5、图2)。从MLGs来看,使用2对引物组合时最大MLGs为65,使用9对引物组合最大MLGs与25对引物的MLGs相同,均为99。
以上结果表明,使用TR23/Psa-04、Psa-03、Psa-05、Psa-06、TR10IGM-1834、TR30Ⅰ、TR1II、Psa-10TR39II、TR64IIGM-1553等9对引物的组合可以达到25对引物的效果,其中TR23和Psa-04可以相互替换,任选其一使用即可。
2.4 引物组合分型效果验证
获得的9对引物组合可将Psa的5种生物型(biovar)准确分开;10株代表性Psa均为biovar 3,其中GZCC7520447和GZCC7520448来自乌当区偏坡乡,独立聚为一支;GZCC7520193和GZCC7520192均来自修文县六桶乡,以相近的遗传距离聚在一起;GZCC7520186和GZCC7520161均来自修文县,以相近的遗传距离聚在一起;biovar 3菌株间存在一定差异(图3)。上述结果表明,所获得的9对引物组合可用于Psa群体遗传结构研究。
3 讨 论
各国学者采用MLVA技术开展Psa群体遗传结构研究共设计了34对MLVA引物[14-16],34对引物中,有些是相同序列(TR14=TR11Ⅱ、TR19=Psa-01);有些是扩增同一TR(TR8与Psa-08、TR39Ⅱ与Psa-10、GM1834与TR10Ⅰ、GM1553与TR64Ⅱ、TR19Psa-01与TR19II);有些序列(TR10I、TR14I、TR15I、TR30I)不能直接被引用,Mazzaglia等[30]的研究中也提到了该不足。有必要针对该34对引物开展系统的分析和筛选,以获得一组可以直接使用、快速精准地对Psa进行分型分析的引物组合。
笔者在本研究中在不降低分辨率的前提下进行MLVA引物组合的筛选,将34对引物减少到只需9对(TR23/Psa-04、Psa-03、Psa-05、Psa-06、TR10IGM-1834、TR30I、TR1II、Psa-10TR39II、TR64IIGM-1553),大大降低了研究的时间和资源成本。该9对引物的组合经验证可准确、快速地对Psa进行分型分析,进行Psa群体遗传结构研究,探索Psa传播和流行规律,为猕猴桃溃疡病防控策略的制定提供科学依据。
笔者在本研究中需查找引物的所有组合并计算SI值,计算量达百万级以上。Optimal Combination Finder (OCF)是一个专门用来找出引物所有可能的组合,并进行引物所有可能组合的SI计算的程序[18]。但由于其操作比较复杂,非开发者很难自行利用该程序进行引物组合的查找和SI的计算,因此笔者基于R开发了一段计算程序,进行所有可能引物组合的查找和SI的计算,它是利用R编写并依托于poppr软件包使用。R语言平台使用方式简单快捷,可多线程运行,提高计算速度,直接采用poppr软件包计算SI值则大大简化了计算步骤,如笔者在本研究中所有引物组合的SI值的计算仅需几个小时便可以完成,这极大地提高了引物组合筛选的效率。该计算程序可以应用于其他微生物MLVA引物的筛选。
笔者在本研究中所引用的34对引物对127株Psa菌株的全基因组扩增信息中,存在部分引物扩增不出条带的情况,但整体来说,供试引物对大多数菌株能扩增出条带,仅Psa-09和TR23引物能扩增出条带的菌株数量较少。不能扩增出条带的原因可能是:(1)菌株序列中本身不存在此位点;(2)GenBank中下载的127株Psa全基因组序列中有118株未组装,未组装的Psa菌株的全基因组信息由几十到几百条序列组成,如果序列截断的位置刚好是位点所在,模拟PCR便无法扩增。无法准确判断扩增不出条带的原因,若将缺失信息的菌株舍弃,样本量将会大大减少,不利于引物组合的筛选。其他的研究者在面对缺失信息时,一般是使用字符将其标记出来,如:Ikawaty等[31]使用“999”表示PCR无扩增信息;Concei??o等[32]使用“99”表示未获得PCR扩增信息;Ciarroni等[14]使用“-1”来表示扩增信息的缺失。笔者在本研究中将缺失信息统一使用“0”表示,一方面便于SI的计算,另一方面也使所有菌株都参与到了引物组合的筛选中,获得了适用于Psa群体遗传结构研究的引物组合。
参考文献 References:
[1] TAKIKAWA Y,SERIZAWA S,ICHIKAWA T,TSUYUMU S,GOTO M. Pseudomonas syringae pv. actinidiae pv. nov.:The causal bacterium of canker of kiwifruit in Japan[J]. Japanese Journal of Phytopathology,1989,55(4):437-444.
[2] SAWADA H,FUJIKAWA T. Genetic diversity of Pseudomonas syringae pv. actinidiae,pathogen of kiwifruit bacterial canker[J]. Plant Pathology,2019,68(7):1235-1248.
[3] 张迪,高小宁,赵志博,秦虎强,黄丽丽. 不同猕猴桃品种对溃疡病的抗性差异及其机制研究[J]. 果树学报,2019,36(11):1549-1557.
ZHANG Di,GAO Xiaoning,ZHAO Zhibo,QIN Huqiang,HUANG Lili. Differences in resistance to Pseudomonas syringae pv. actinidiae and acting mechanism of different kiwifruit varieties[J]. Journal of Fruit Science,2019,36(11):1549-1557.
[4] 虞江,李渺,张文娟,冯双,卢永仲,徐志华,张善淇,龚子雄,何鹏,魏娴. 贵州省贵阳市修文县猕猴桃溃疡病发生现状调查[J]. 贵州农机化,2022(1):41-43.
YU Jiang,LI Miao,ZHANG Wenjuan,FENG Shuang,LU Yongzhong,XU Zhihua,ZHANG Shanqi,GONG Zixiong,HE Peng,WEI Xian. Investigation on the current situation of kiwifruit canker disease in Xiuwen County,Guiyang City,Guizhou Province[J]. Guizhou Agricultural Mechaniation,2022(1):41-43.
[5] 马利. 四川猕猴桃溃疡病发生区划和综合防控技术研究[D]. 雅安:四川农业大学,2018.
MA Li. Occurrence regionalization and integrated management of kiwifruit canker caused by Pseudomonas syringae pv. actinidiae in Sichuan[D]. Yaan:Sichuan Agricultural University,2018.
[6] 王茹琳,刘原,李庆,沈沾红,陆兴利,赵金鹏,王闫利,王明田. 气候变化情景下四川省猕猴桃溃疡病菌潜在地理分布模拟[J]. 植物保护,2020,46(2):38-47.
WANG Rulin,LIU Yuan,LI Qing,SHEN Zhanhong,LU Xingli,ZHAO Jinpeng,WANG Yanli,WANG Mingtian. Analysis of geographical distribution of Pseudomonas syringae pv. actinidiae in Sichuan under climate change[J]. Plant Protection,2020,46(2):38-47.
[7] FUJIKAWA T,SAWADA H. Draft genome sequences of nine Japanese strains of the kiwifruit bacterial canker pathogen Pseudomonas syringae pv. actinidiae biovar 3[J]. Microbiology Resource Announcements,2020,9(45):e01007-e01020.
[8] 朱海云,马瑜,柯杨,李勃. 陕西省猕猴桃溃疡病病原菌分离鉴定及分型研究[J]. 微生物学杂志,2023,43(4):74-83.
ZHU Haiyun,MA Yu,KE Yang,LI Bo. Isolation,identification and typing the pathogen of Chinese gooseberry or kiwifruit (Actinidia chinensis) canker in Shaanxi Province[J]. Journal of Microbiology,2023,43(4):74-83.
[9] HE R,LIU P,JIA B,XUE S Z,WANG X J,HU J Y,AL SHOFFE Y,GALLIPOLI L,MAZZAGLIA A,BALESTRA G M,ZHU L W. Genetic diversity of Pseudomonas syringae pv. actinidiae strains from different geographic regions in China[J]. Phytopathology,2019,109(3):347-357.
[10] 代玉立,兰成忠,甘林,刘晓菲,龚国淑,杨秀娟. 中国4省猕猴桃细菌性溃疡病菌的生物型检测和遗传多样性分析[J]. 植物保护,2022,48(6):58-68.
DAI Yuli,LAN Chengzhong,GAN Lin,LIU Xiaofei,GONG Guoshu,YANG Xiujuan. Detection of Pseudomonas syringae pv. actinidiae biovars from four provinces in China and genetic diversity analysis[J]. Plant Protection,2022,48(6):58-68.
[11] VAN BELKUM A. Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA)[J]. FEMS Immunology & Medical Microbiology,2007,49(1):22-27.
[12] OSANLOO L,ZEIGHAMI H,HAGHI F,SHAPOURI R,SHOKRI R. Molecular typing of multidrug-resistant Acinetobacter baumannii isolates from clinical specimens by ERIC-PCR and MLVA[J]. Current Microbiology,2023,80(11):355.
[13] SIARKOU V I,VORIMORE F,VICARI N,MAGNINO S,RODOLAKIS A,PANNEKOEK Y,SACHSE K,LONGBOTTOM D,LAROUCAU K. Diversification and distribution of ruminant Chlamydia abortus clones assessed by MLST and MLVA[J]. PLoS One,2015,10(5):e0126433.
[14] CIARRONI S,GALLIPOLI L,TARATUFOLO M C,BUTLER M I,POULTER R T M,POURCEL C,VERGNAUD G,BALESTRA G M,MAZZAGLIA A. Development of a multiple loci variable number of tandem repeats analysis (MLVA) to unravel the intra-pathovar structure of Pseudomonas syringae pv. actinidiae populations worldwide[J]. PLoS One,2015,10(8):e0135310.
[15] CUNTY A,CESBRON S,POLIAKOFF F,JACQUES M A,MANCEAU C. Origin of the outbreak in France of Pseudomonas syringae pv. actinidiae biovar 3,the causal agent of bacterial canker of kiwifruit,revealed by a multilocus variable-number tandem-repeat analysis[J]. Applied and Environmental Microbiology,2015,81(19):6773-6789.
[16] 赵志博,杜淑凤,李月,杨文,樊荣,王勇,龙友华. 猕猴桃溃疡病菌biovar 3群体MLVA分型技术的建立与应用[J]. 植物病理学报,2019,49(4):445-455.
ZHAO Zhibo,DU Shufeng,LI Yue,YANG Wen,FAN Rong,WANG Yong,LONG Youhua. Establishment and application of MLVA typing for Pseudomonas syringae pv. actinidiae biovar 3 populations[J]. Acta Phytopathologica Sinica,2019,49(4):445-455.
[17] MCCANN H C,LI L,LIU Y F,LI D W,PAN H,ZHONG C H,RIKKERINK E H A,TEMPLETON M D,STRAUB C,COLOMBI E,RAINEY P B,HUANG H W. Origin and evolution of the kiwifruit canker pandemic[J]. Genome Biology and Evolution,2017,9(4):932-944.
[18] 赵志博. 猕猴桃细菌性溃疡病菌群体结构与致病机制研究[D]. 杨凌:西北农林科技大学,2016.
ZHAO Zhibo. Population composition and pathogenic mechanism in Pseudomonas syringae pv. actinidiae[D]. Yangling:Northwest A & F University,2016.
[19] MAZZAGLIA A,STUDHOLME D J,TARATUFOLO M C,CAI R M,ALMEIDA N F,GOODMAN T,GUTTMAN D S,VINATZER B A,BALESTRA G M. Pseudomonas syringae pv. actinidiae (PSA) isolates from recent bacterial canker of kiwifruit outbreaks belong to the same genetic lineage[J]. PLoS One,2012,7(5):e36518.
[20] SAWADA H,MIYOSHI T,IDE Y. Novel MLSA group (Psa5) of Pseudomonas syringae pv. actinidiae causing bacterial canker of kiwifruit (Actinidia chinensis) in Japan[J]. Japanese Journal of Phytopathology,2014,80(3):171-184.
[21] FUJIKAWA T,SAWADA H. Genome analysis of Pseudomonas syringae pv. actinidiae biovar 6,which produces the phytotoxins,phaseolotoxin and coronatine[J]. Scientific Reports,2019,9:3836.
[22] MARCELLETTI S,FERRANTE P,PETRICCIONE M,FIRRAO G,SCORTICHINI M. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species[J]. PLoS One,2011,6(11):e27297.
[23] PAN X,ZHAO S Y,WANG Y Z,LI M Z,HE L Q,ZHUANG Q G. Complete genome sequencing of Pseudomonas syringae pv. actinidiae Biovar 3, P155, kiwifruit pathogen originating from China[J]. Bioscience Journal,2020,36(6):2220-2228.
[24] FUJIKAWA T,SAWADA H. Genome analysis of the kiwifruit canker pathogen Pseudomonas syringae pv. actinidiae biovar 5[J]. Scientific Reports,2016,6:21399.
[25] MCCANN H C,RIKKERINK E H A,BERTELS F,FIERS M,LU A,REES-GEORGE J,ANDERSEN M T,GLEAVE A P,HAUBOLD B,WOHLERS M W,GUTTMAN D S,WANG P W,STRAUB C,VANNESTE J L,RAINEY P B,TEMPLETON M D. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease[J]. PLoS Pathogens,2013,9(7):e1003503.
[26] CAO Y F,WANG L J,XU K X,KOU C H,ZHANG Y L,WEI G F,HE J J,WANG Y F,ZHAO L P. Information theory-based algorithm for in silico prediction of PCR products with whole genomic sequences as templates[J]. BMC Bioinformatics,2005,6:190.
[27] WANG X,HUANG B X,BLAIR B,EGLEZOS S,BATES J. Selection of optimal combinations of loci by the Optimal Combination Finder computer program from a group of variable number tandem repeat loci for use in Staphylococcus aureus food poisoning case investigations[J]. Journal of Medical Microbiology,2012,61(Pt 5):631-639.
[28] R Core Team. R: A language and environment for statisticalcomputing[CP]. R Foundation for Statistical Computing,Vienna,Austria. URL. 2020. https://www.R-project.org/.
[29] KAMVAR Z N,TABIMA J F,EVERHART S E,BROOKS J C,KRUEGER-HADFIELD S A. Package ‘poppr[CP]. 2020. https://grunwaldlab.github.io/poppr.
[30] MAZZAGLIA A,TURCO S,TARATUFOLO M C,TAT? M,RAHI Y J,GALLIPOLI L,BALESTRA G M. Improved MLVA typing reveals a highly articulated structure in Pseudomonas syringae pv. actinidiae populations[J]. Physiological and Molecular Plant Pathology,2021,114:101636.
[31] IKAWATY R,WILLEMS R J L,BOX A T A,VERHOEF J,FLUIT A C. Novel multiple-locus variable-number tandem-repeat analysis method for rapid molecular typing of human Staphylococcus aureus[J]. Journal of Clinical Microbiology,2008,46(9):3147-3151.
[32] CONCEI??O T,DE SOUSA M A,DE LENCASTRE H. Staphylococcal interspersed repeat unit typing of Staphylococcus aureus:Evaluation of a new multilocus variable-number tandem-repeat analysis typing method[J]. Journal of Clinical Microbiology,2009,47(5):1300-1308.
收稿日期:2023-10-19 接受日期:2023-11-09
基金项目:贵州省科技计划项目(黔科合基础[2019]1308号)
作者简介:姚令,男,研究实习员,硕士,研究方向为植物病理学。E-mail:1158142736@qq.com
*通信作者 Author for correspondence. E-mail:gzusp@126.com
猜你喜欢
快乐语文(2021年36期)2022-01-18 05:48:38
基层中医药(2021年2期)2021-07-23 01:41:48
动漫星空(兴趣百科)(2020年12期)2020-12-12 05:31:38
基层中医药(2020年5期)2020-09-11 06:32:00
作文小学中年级(2019年9期)2019-10-14 00:39:48
今日农业(2019年14期)2019-01-04 08:57:40
基层中医药(2018年5期)2018-08-31 02:35:42
现代园艺(2018年3期)2018-02-10 05:18:39
创新作文(小学版)(2016年23期)2016-12-01 05:51:04
制造技术与机床(2015年10期)2015-04-09 07:06:14
