
基于稀疏自编码器的细胞类型反卷积模型SMCTD 设计与实现
2024-06-03 07:59:45朱贤振李雪玲
电脑知识与技术 2024年11期
关键词:深度学习
朱贤振 李雪玲
摘要:单细胞 RNA 测序已成为研究生物学重要特征的强大高分辨率工具。然而,其测序条件苛刻,价格成本高昂。目前细胞类型反卷积能够很好地解决这些限制问题,SMCTD(Sparse Model Cell Type Deconvolution) 使用稀疏自编码器优化TAPE(Tissue-AdaPtive autoEncoder) ,使其在直肠癌和PBMC模拟数据上预测细胞类型比列具有更高的灵敏度、准确性和整体性能,同时在预测细胞类型特异性基因表达上表现更优。
关键词:单细胞测序;细胞类型反卷积;深度学习;稀疏自编码器;一致性相关系数
中图分类号:TP311 文献标识码:A
文章编号:1009-3044(2024)11-0009-04
随着二代深度测序(NGS) 、单细胞转录组测序(scRNA-seq) 、空间转录组(Spatial Transcriptomics) 技术、细胞类型反卷积算法的发展,为整合单细胞测序数据和大规模 bulk 基因表达谱,研究癌症微环境中的细胞组成和基因表达,提供了重要手段。然而,现有的细胞类型反卷积方法[1]的准确度和解析颗粒度有很大提升空间,发展基于深度机器学习的细胞类型反卷积算法,能够实现并加速大范围内高通量临床数据的精确分析[2]。
TAPE[3]是一种连接批量RNA-seq和单细胞RNAseq[4]的深度学习方法,可在短时间内实现精确的反卷积。通过构建可解释的解码器并在独特的方案下进行训练,TAPE 可以自适应地预测细胞类型分数和细胞类型特异性基因表达。与多个数据集上的流行方法相比,TAPE 在细胞类型水平上具有更好的整体性能和相当的准确性。此外,它在不同细胞类型中更稳健、更快、更灵敏,可以提供具有生物学意义的预测。然而TAPE是基于传统自编码器的深度学习模型,需要对所有数据进行处理降维,容易过拟合,尽管传统自编码器可以学习数据的特征表示,但并不保证这些特征是有意义、可解释的。模型可能会学习到捕捉数据中的一些噪声或冗余信息,而不是真正有用的特征。
稀疏自编码器相比自编码器特征的学习与选择能力更好,稀疏自编码器通过强制编码层的神经元保持较低的激活率,鼓励网络只激活最重要的特征神经元[5]。这可以使更有意义、更具区分性的特征被学习和保留,从而提高模型对数据的表征能力。也能降低过拟合的风险: 通过在编码层施加稀疏性约束,稀疏自编码器可以降低过拟合的风险。这是因为模型被迫仅仅选择最重要的特征,而不会过度适应训练数据中的噪声或不重要的变化。更好的泛化能力: 由于稀疏自编码器倾向于学习更有意义的特征,它们通常能够更好地泛化到新的、未见过的数据,从而提高模型的泛化能力。还拥有更高效的特征表示:稀疏自编码器可以学习数据的更紧凑、更高级的表示。这种表示可以更好地捕捉数据中的关键模式和结构,从而在后续的任务(如分类、聚类等)中表现更好。
1 细胞类型反卷积模型构建
细胞类型反卷积是能够将大规模bulk基因表达谱反卷积得到bulk中细胞类型比例和细胞类型特异性基因的一种方法,传统细胞类型反卷积需要制作签名矩阵,签名矩阵是基于一组特定基因的表达模式构建的。这些特定基因通常与某个生物学特性、状态或功能相关联。通过分析大量单细胞数据,可以从中提取出这些特定基因的表达模式,并将这些模式整合到一个矩阵中,即签名矩阵[1]。非常烦琐,最近几年兴起的深度学习细胞类型反卷积能够自主学习bulk基因表达谱中特征,无须构建签名矩阵即可进行反卷积。这些方法中,TAPE是反卷积性能最出色的之一,比同样使用深度学习的Scaden [2]更稳健,与传统细胞类型反卷积方法CIBERSORTx [1]性能旗鼓相当。而TAPE 使用自编码器的缺点也让我们思考用更优秀的网络构建模型来提升性能。
1.1 模型设置
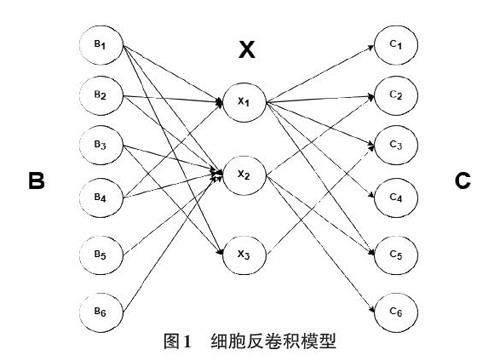
如图1所示,我们引入稀疏自编码器,与传统自编码器相比,引入稀疏性目标值和稀疏性权重,稀疏性目标值是一个预设的小数,表示隐藏层神经元的期望平均激活度,通常是0.05或者更小。稀疏性权重是一个常数,表示稀疏性惩罚项在总代价函数中的权重,用来控制稀疏性的强度。图1中B 表示输入的bulk基因表达数据,C 表示经过稀疏自编码器编码再解码重现的bulk基因表达数据,X 表示通过编码器得到的细胞类型比例。图1左半部分表示为编码器,是一个回归模型,负责将高维bulk基因表达数据映射到低维的细胞类型比例数据。相反,图1右半部分可以根据细胞类型比例数据重建bulk基因表达数据。
1.2 模型训练
我们先预设稀疏性目标值和稀疏性权重,稀疏性目标值代表期望的神经元平均激活度,稀疏性权重用来控制稀疏性强度。然后在代码中定义了KL散度函数(Kullback-Leibler Divergence) ,使用KL散度函数和稀疏性目标值和权重计算稀疏性惩罚损失[5]。
我们使用大约5 000个bulk样本进行训练。使用预测细胞类型比例和真实细胞类型比例之间的MAE (平均绝对误差)与稀疏性惩罚损失的和来优化编码器的参数,并使用重构bulk数据和原始bulk数据之间的MAE 与稀疏性惩罚损失的和来优化解码器和编码器。
1.3 模型預测
使用反卷积模型进行预测需要预先准备单细胞参考数据,行为细胞类型,列为基因名称,文件为TXT 格式。为了使反卷积结果更精准更具有生物学意义,单细胞参考数据要与需要预测的bulk 数据为同一组织的,并且拥有相同的细胞类型。
需要预测的bulk 数据需要指定分隔符,行为样本名称,列为基因名称,数据类型最好为“counts”,若使用“TPM”或者“FPKM”格式须自备基因长度文件使数据最终以counts格式运行在程序上。
我们把准备好的单细胞参考数据和bulk数据输入程序,将模式选择为“overall”,然后选择合适的数据类型及基因长度文件,自适应参数选择为“True”或者“False”。如果是“ True”,那么它将会预测输出签名矩阵,反之,则返回空值。等待程序运行完成,会得到预测的细胞类型比例数据(行为样本,列为细胞类型)及选择可得到的签名矩阵。
2 细胞类型反卷积模型性能比较
由于公共数据库中同一样本中既测bulk数据又测单细胞数据的少之又少,所以为了精准测出反卷积模型的性能,因此有必要进行伪bulk数据测试进行估计。伪bulk数据是通过具有基本事实(预定义的细胞类型比例)的单细 胞基因表达数据在计算机中生成的。也就是说,伪bulk数据是许多单细胞基因表达数据的总和。我们将使用TAPE中的伪bulk模拟程序模拟bulk。
2.1 GSE176078单细胞数据模拟预测比较
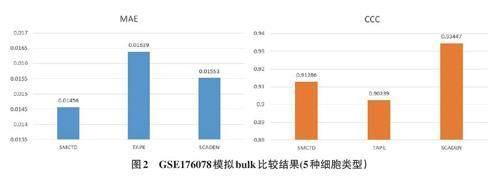
首先,我们预设真实细胞类型比例,再将从GEO 中下载的乳腺癌数据集GSE176078 [6]的样本作为参考的单细胞数据生成模拟bulk对五种细胞类型进行反卷积性能预测。设定两个参数指标:MAE(平均绝对误差)和CCC(一致性相关系数)[7],MAE是对每个细胞类型的预测值与其对应的实际值之间的绝对差值进行求和,然后对所有数据点的绝对差值求平均值,数值越小性能越好。CCC是评价细胞类型比例预测值与真实值之间的一致性的指标,CCC值越接近1代表性能越好。
最终结果如图2所示,我们的模型SMCTD(稀疏自编码器)在误差方面要比TAPE和Scaden都低,在CCC方面也要比TAPE更出色,略逊于Scaden,综合两方面来看,SMCTD是三者中反卷积性能最出色的。
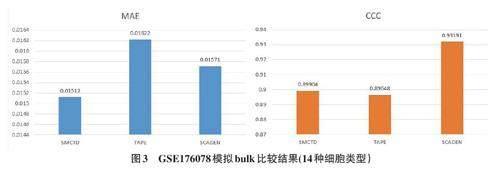
接下来测试模型在细胞类型增加的情况下的性能,我们继续用GSE176078模拟bulk,这次bulk包含14 种细胞亚型,例如:Monocyte、Fibroblasts、NK cells 等。評价指标同上。
如图3所示,结果表明,在这种情况下,所有方法都出现性能下降的情况,但这些方法的MAE与预测五种细胞类型情形中的 MAE相当,这表明这些方法可以预测接近真实值的值。同时SMCTD也是这种情况下误差最低的算法,其CCC值也为三种方法第二高,说明其性能是三者间最出色的。
2.2 PBMC 单细胞数据模拟预测比较
接下来我们使用10X Genomics官网的PBMC(外周血单个核细胞)单细胞数据[8]模拟bulk(其中包含七种细胞类型)进行反卷积,评价指标同上。
最终结果如图4 所示,可以发现在PBMC 数据上Scaden 表现是最出色的,我们的模型SMCTD 虽然比Scaden 略差,但要比TAPE在MAE、CCC值两个方面都要更优秀。
综上所述,我们发现模型SMCTD在预测细胞类型方面有着不逊于TAPE、Scaden的性能,虽然这三种方法在细胞类型增多的情况下都会出现性能下降,这种情况是可以预见的,所以解决这一问题也是未来研究的方向之一。
2.3 组织适应性细胞类型特异性基因表达预测
SMCTD不仅可以预测细胞类型分数,同样可以自适应地预测细胞类型特异性基因表达。也就是说,SMCTD 只需要模拟数据来训练,如果给出相应的bulk RNAseq数据,它可以预测细胞类型特异性的基因表达。此功能使SMCTD 能够剖析不同细胞类型中的bulk基因表达,并发现不同细胞类型中一些潜在差异表达的基因。
我们测试了预测的细胞类型特异性 GEP 的正确性。为了测试这一点,我们测量了每种细胞类型的预测基因表达值与从单细胞RNA-seq获得的原始基因表达值之间的一致性(图5) 。这里,bulk 数据用GSE176078单细胞数据模拟生成,而单细胞数据是乳腺癌癌单数据集GSE176078。由于在训练阶段使用Log2 和 MinMaxScaler() 将输入的 RNA-seq 数据转换为 0-1 值,因此按细胞类型分组的基因表达值的总和也以这种方式转换以进行比较与预测的相对基因表达值。
由图5、图6和表1可得,SMCTD在四种细胞类型上的基因表达预测一致性相关系数都要高于TAPE,仅在免疫细胞中表现不佳,考虑到其他四种细胞类型的良好一致性,这种失真可能是由个体差异引起的。图表中显示的一致性证明SMCTD正确预测了细胞类型特异性基因表达,为进一步的基因表达分析奠定了基础。
3 结束语
本文优化了一个细胞类型反卷积模型TAPE,使用了稀疏自编码器作为模型基础,增强了模型的稀疏性,提高了模型性能,使细胞类型反卷积模型的误差降低,且提高了相关性,在我们测试的两类数据和多种细胞类型上均能体现出。此外,在预测细胞类型特异性基因表达上SMCTD也比TAPE在大多数细胞类型上的结果更准确。
但模型在细胞类型过多的情况下表现下降,这是未来要攻克的方向之一。在预测细胞类型特异性基因表达方面,SMCTD同样有一定的优化上升空间。
参考文献:
[1] NEWMAN A M, STEEN C B, LIU C L, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry [J]. Nat Biotechnol, 2019, 37(7): 773.
[2] MENDEN K, MAROUF M, OLLER S, et al. Deep learningbased cell composition analysis from tissue expression profiles[J]. Science Advances,2020,6(30):eaba2619.
[3] CHEN Y S,WANG Y X,CHEN Y L,et al. Deep autoencoder for interpretable tissue-adaptive deconvolution and cell-typespecific gene analysis[J]. Nature Communications,2022,13(1):6735.
[4] WANG Z,GERSTEIN M,SNYDER M. RNA-Seq:a revolutionary tool for transcriptomics[J]. Nature Reviews Genetics,2009,10(1):57-63.
[6] WU S Z,AL-ERYANI G,RODEN D L,et al. A single-cell and spatially resolved atlas of human breast cancers[J]. Nature Genetics,2021,53(9):1334-1347.
[5] NG A. Sparse autoencoder[R]. CS294A Lecture Notes, 2011:72.
[6] WU S Z,AL-ERYANI G,RODEN D L,et al. A single-cell and spatially resolved atlas of human breast cancers[J]. Nature Genetics,2021,53(9):1334-1347.
[7] LIN L I. A concordance correlation coefficient to evaluate reproducibility[J]. Biometrics,1989,45(1):255-268.
[8] CHEMISTRY) K P F A H D V. single cell gene expression dataset by cell ranger 2. 1. 0 [DS]. 10X Genomics, 2017,
【通联编辑:李雅琪】
猜你喜欢
中国教育技术装备(2016年19期)2016-12-27 19:23:52
中国远程教育(2016年11期)2016-12-27 18:07:31
现代商贸工业(2016年25期)2016-12-26 09:58:02
江苏教育·中学教学版(2016年11期)2016-12-21 11:45:08
江苏教育·中学教学版(2016年11期)2016-12-21 11:36:29
现代情报(2016年10期)2016-12-15 11:50:53
考试周刊(2016年94期)2016-12-12 12:15:04
新教育时代·教师版(2016年23期)2016-12-06 06:02:38
法制与社会(2016年32期)2016-12-01 15:25:53
软件导刊(2016年9期)2016-11-07 22:20:49
