
一株罗非鱼无乳链球菌Streptococcus agalactiae CS2108的全基因组测序及功能分析
2024-05-25 13:03施金谷马振花王志强韩书煜罗福广文衍红苏在权佀再勇
南方农业 2024年5期
施金谷,马振花,王志强,韩书煜,罗福广,文衍红,易 弋,苏在权,佀再勇*
(1.广西壮族自治区水产技术推广站,广西南宁 530199;2.广西科技大学生物与化学工程学院,广西柳州 545006;3.柳州市渔业技术推广站,广西柳州 545006)
罗非鱼主要分布在热带、亚热带地区,对环境适应能力强,生长速度快,繁殖能力强,并且其肉味鲜美、肉质细嫩、食用方法简单、营养丰富,深受广大养殖户的青睐[1-2]。发展罗非鱼养殖产业对于增加农民就业机会和收入、促进国际贸易具有十分重要的意义[3-4]。
随着罗非鱼养殖模式向着高密度、集约化养殖发展,加上养殖水质、环境及气候的恶化,渔民防控意识的欠缺,导致各类病害常大面积暴发[5]。其中链球菌病是罗非鱼健康养殖的最大威胁,最主要的病原菌是无乳链球菌(Sreptococcus agalactiae)。近年来,罗非鱼无乳链球菌病在我国广东、广西和海南等地时有发生,具有暴发范围广、死亡率高的特点[6]。无乳链球菌大多呈球形,链状排列,革兰氏染色阳性,具有荚膜和鞭毛,无芽孢,适宜在温度28~37 ℃、pH 值7.4~7.6 条件下生长。在脑心浸液琼脂固体培养基上呈卵圆形,菌落表面光滑。目前,利用Lance-field 法可以将其分成Ia、Ib、Ⅱ~Ⅸ共10 个血清型[7]。该病主要病理特征为眼球突出或浑浊发白[8],眼眶充血,腹部膨大,肛门红肿,少食或者绝食,游姿平衡失调,解剖病鱼可见胆囊肿大,胆汁稀薄,色浅,肠内充满淡黄色液体,肝脏增大[9-10]。组织病理学表现为鳃充血,肾脏受损、肿大、白细胞浸润,肝脏颗粒变性,肠道粘膜上皮变性、坏死、脱落、固有膜上炎性白细胞浸润,眼睛脉络膜和眶骨膜组织炎性坏死等[11]。
目前细菌性疾病的诊断方法主要有生理生化鉴定、免疫学方法、分子生物学技术[12]。分子生物学诊断技术中常用的方法有PCR 技术、核酸分子杂交、多位点序列分型、环介导等温扩增技术等。王均等基于罗非鱼无乳链球菌的cfb和16S rRNA 基因序列建立了快速检测无乳链球菌的双重PCR 方法[13];樊海平等根据罗非鱼无乳链球菌16S rRNA 基因和sip基因序列用于罗非鱼无乳链球菌病的流行病学调查[14],这些分子生物学诊断方法都离不开无乳链球菌的基因组信息。
截至目前,NCBI 数据库上有1 867 个无乳链球菌基因组信息,这些无乳链球菌基因组序列中人源和鱼源的比较多,部分是牛源、蛙源和犬源的。前期实验室从患病罗非鱼中分离出1 株无乳链球菌,命名为S.agalactiaeCS2108,为深入了解该罗非鱼无乳链球菌S.agalactiaeCS2108 的全基因组序列,分析该菌的基因组基本特征,为后续罗非鱼无乳链球病的防治提供理论支撑。本研究采用Illumina Hiseq+PacBio 的测序方式对S.agalactiaeCS2108 菌株进行全基因组测序分析,并进行组装和注释、毒力基因和耐药基因的预测,分析和预测结果将为罗非鱼无乳链球菌的预防和防治提供理论支持。
1 材料与方法
1.1 菌株培养与基因组DNA提取
无乳链球菌S.agalactiaeCS2108 分离于患病的罗非鱼肝脏组织,保存于广西科技大学生物与化学工程学院。将S.agalactiaeCS2108 从甘油管接种到脑心浸液培养基(Brian Heart Infusion,BHI)固体培养基平板上,在37 ℃培养箱中过夜培养。挑取单菌落并接种至250 mL的BHI液体培养基中,在温度37 ℃、转速200 rpm的摇床中培养到对数期。然后在4 ℃、12 000 rpm 条件下离心10 min,收集菌体,送至上海美吉生物医疗科技有限公司进行全基因组测序。
1.2 系统进化树的构建
基于S.agalactiaeCS2108 的16S rRNA 序列,通过与NCBI 数据库对比,选择在种属水平上最接近的20株菌的16S rRNA 序列,通过MEGA 6.0 软件选择邻近(Neighbor-Joining)法构建系统进化树。
1.3 文库构建与测序
基因组测序使用Nanopore PromethION 和Illumina NovaSeq 6000测序平台进行全基因组测序。Illumina测序数据被用来评估基因组的杂合度、重复度,评估基因组大小,以及是否存在质粒及污染,辅助后续组装策略的选择。同时由于三代测序具有较高的测序错误率,用二代测序数据对三代数据进行校正,保证组装结果具有较高的准确度。
取不少于1 μg 基因组DNA,利用Covaris 进行基因组DNA 片段化,将DNA 样本剪切成约400 bp 的片段,琼脂糖凝胶鉴定片段大小、分布,富集300~500 bp范围的基因组片段,使用NEXTflex™Rapid DNA-Seq试剂盒进行文库制备。具体步骤如下:连接A&B 接头;去除接头自连片段;琼脂糖凝胶电泳进行片段筛选,保留一端是A 接头、一端是B 接头的片段;氢氧化钠变性,产生单链DNA片段;桥式PCR扩增。
制备的文库在Illumina Novaseq 6000 仪器上进行双端测序(2×150 bp)。具体步骤如下:加入改造过的DNA聚合酶和带有4种荧光标记的dNTP,每次循环只掺入单种碱基;用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类;将“荧光基团”和“终止基团”化学切割,恢复3′端粘性,继续聚合第二个核苷酸;统计每轮收集到的荧光信号结果,获知模板DNA 片段的序列。Nanopore 测序是一种纳米孔测序技术。具体步骤如下:在测序前,双链DNA 末端需添加2 个携带运动蛋白的接头(引导接头和发夹接头)。当测序开始,引导接头(leader adapter)将双链DNA 片段引导至纳米孔附近,引导运动蛋白将双链DNA 解螺旋成单链,使第一链(模板链)穿过纳米孔。在模板链的末端,发夹运动蛋白再以类似的方式介导互补链通过纳米孔,从而保证DNA的双链测序。当DNA 链穿过纳米孔时,传感器以恒定的频率测量离子电流变化,根据电流变化的频谱,应用模式识别算法得到碱基序列。
1.4 基因组组装、基因预测与注释
利用Nanopore PromethION 和Illumina NovaSeq6000平台生成的数据进行生物信息学分析。所有分析均在上海美吉生物云(http://cloud.majorbio.com)上进行。具体程序如下:Illumina 测序下机的原始数据(raw data)以Fast 格式储存,为了使后续的组装更加准确,使用软件Fast v0.23.0 对原始数据进行质量剪切。利用Unicycler v0.4.8[15]对质控之后的Illumina 数据和Nanopore 数据进行混合组装,最后使用Pilon v1.22 将Illumina 短序列mapping 到组装好的基因组上进行矫正。
利用Prodigal v2.6.3[16]对基因组中的编码序列(CDS)进行预测,质粒基因采用GeneMarkS[17]软件预测,tRNAscan-SE v2.0[18]进行tRNA 预测,Barrnap v0.9(https://github.com/tseemann/barrnap) 进 行rRNA 预 测。利用BLASTP、Diamond、HMMER 等序列比对工具,从NR、Swiss-Prot、Pfam、GO、COG、KEGG 数据库中对预测到的CDS进行功能注释。
1.5 毒力和耐药基因预测
将无乳链球菌S.agalactiaeCS2108 基因组信息比对毒力因子数据库VFDB (Version: 2016.03,http://www.mgc.ac.cn/VFs/main.htm)获得毒力因子基因注释概况并进行统计;通过耐药基因数据库(CARD Version 1.1.3,DOI: 10.1128/AAC.00419-13)获得每个基因组中包含的耐药基因的信息。
2 结果与分析
2.1 S.agalactiae CS2108的16S rRNA进化树分析
基于菌株S.agalactiaeCS2108 16S rRNA 基因序列与NCBI 数据库中相似菌株序列比对结果显示无乳链球菌菌株CS2108 和GCF_000186445.1 相似度最高(见图1)。
2.2 基因组组装与注释
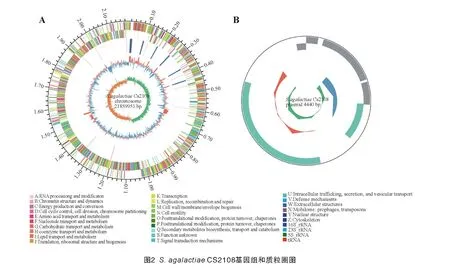
原始测序数据结果表明,无乳链球菌S.agalactiaeCS2108 基因组包含1 条染色体和1 个质粒,染色体大小为2 189 951 bp,GC 含量为35.82%;质粒大小为4 440 bp,GC含量为37.82%。基因组共编码2 110个基因,所有编码基因总长度为1 922 967 bp,基因平均长度911.36 bp,占整个基因组总长度的96%,含有80个tRNA、21个rRNA(5S RNA、16S RNA和23S RNA 各7 个);含有8 个基因岛,基因岛基因总长度为163 023 bp,平均每个基因岛长20 377.88 bp,8个基因岛共有基因179个,平均每个基因岛有22.38个基因。包含2 个前噬菌体,一个位于622685~672418处,序列总长度为49 734 bp,包含47个基因,另一个位于2135280~2145186 处,序列总长度为9 907 bp,包含17 个基因。含有11 个CRISPR-Cas,共含有重复序列107 个,平均重复序列长度35.67 bp(见表1)。Circos 基因组圈图可以全面展示基因组的特征,从外到内圆圈对应的信息依次为基因组大小标识、正链、负链上的基因信息、ncRNA、GC 含量、GC-Skew。菌株S.agalactiaeCS2108 的基因组圈图见图2A(见封三),质粒圈见图2B(见封三),基因组序列登录号分别为CP123969、CP123970。

表1 无乳链球菌S.agalactiae CS2108基因组特征
2.3 基因组功能分析
2.3.1 GO注释结果
菌株S.agalactiaeCS2108共有1 640个基因注释到GO 功能,占基因组的77.73%,结果如图3(见封三),其中与生物过程相关的基因有843个,与细胞组成相关的基因有807 个,与分子功能相关的基因有1 369 个。在生物过程中,与转录和翻译相关的基因排前2 位(共109 个),在细胞组分中,排前2 位的基因是与质膜和细胞质成分相关的基因(共642 个);在分子功能方面,排前2 位的基因是与ATP 结合、DNA 结合相关的基因(共440个)。

2.3.2 COG注释结果
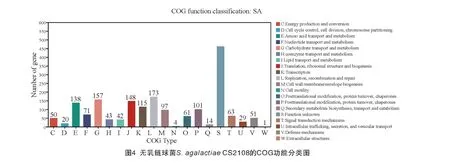
菌株S.agalactiaeCS2108共有1 820个基因注释到COG 功能,占基因组的86.26%,结果如图4(见封三),其中有462个未知功能的基因,功能已知的基因有173个与复制、重组和修复相关,有157 个基因与碳水化合物运输和代谢相关,有148 个基因与翻译、核糖体结构和生物发生相关,有138 个基因与氨基酸运输和代谢相关,有115个基因与转录相关。
2.3.3 KEGG注释结果
菌株S.agalactiaeCS2108共有1 213个基因注释到功能信息,占基因组的57.49%,结果如图5(见封三),参与细胞过程的基因有72个,参与环境信息处理的基因有197个,参与遗传信息处理的基因有157个,参与人类疾病的基因有68 个,参与代谢的基因有959 个,参与生物系统的基因有35 个。在KEGG 代谢通路中代谢相关的基因最多,在代谢途径中排前三的分别是全局图与概览图、碳水化合物代谢和氨基酸代谢。

2.3.4 毒力因子预测
将菌株S.agalactiaeCS2108 的基因组与毒力因子数据库(Virulence factors database,VFDB)进行比对,得到毒力因子基因注释的概况,结果表明有242 个毒力因子基因得到注释,分别是68个防御性毒力因子基因(Defensive virulence factors)(图6-A)、48 个非特异性毒力因子基因(Nonspecific virulence factor)(图6-B)、88 个攻击性毒力因子基因(Offensive virulence factors)(图6-C)和20个毒力相关的调控基因(图6-D)。在防御性毒力因子基因中,最多的是抗吞噬基因有30 个,其次是血清抗性相关的基因22 个,胁迫蛋白相关的基因11个,最少的是补充蛋白酶基因1 个;在非特异性毒力因子中,最多的是铁离子吸收系统基因有42个,胞外酶和镁离子吸收相关基因分别有3 个;在防御性毒力因子基因中,最多的是毒素相关的基因有36 个,粘附相关的基因有31 个,分泌系统和侵染相关的基因分别是14 个和7 个;毒力相关的调控基因有20个。由上可知,影响毒力的因素是多类基因相互协调共同作用的结果。

图6 S.agalactiae CS2108的毒力因子统计
2.3.5 耐药基因预测
将菌株S.agalactiaeCS2108 的基因组与综合的抗生素抗性基因数据库(Comprehensive Antibiotic Resistance Database,CARD)进行比对,获得基因组中包含耐药基因的信息。由表2 可见,S.agalactiaeCS2108 的基因组中含有耐药基因141 个,排在前五的耐药基因是46 个大环内酯类抗生素基因(Macrolide antibiotic)、29 个四环素抗生素基因(Tetracycline antibiotic)、25 个氟喹诺酮类抗生素基因(Fluoroquinolone antibiotic)、17 个盐酸克林霉素基因(Lincosamide antibiotic)和16 个糖肽抗生素基因(Glycopeptide antibiotic)。
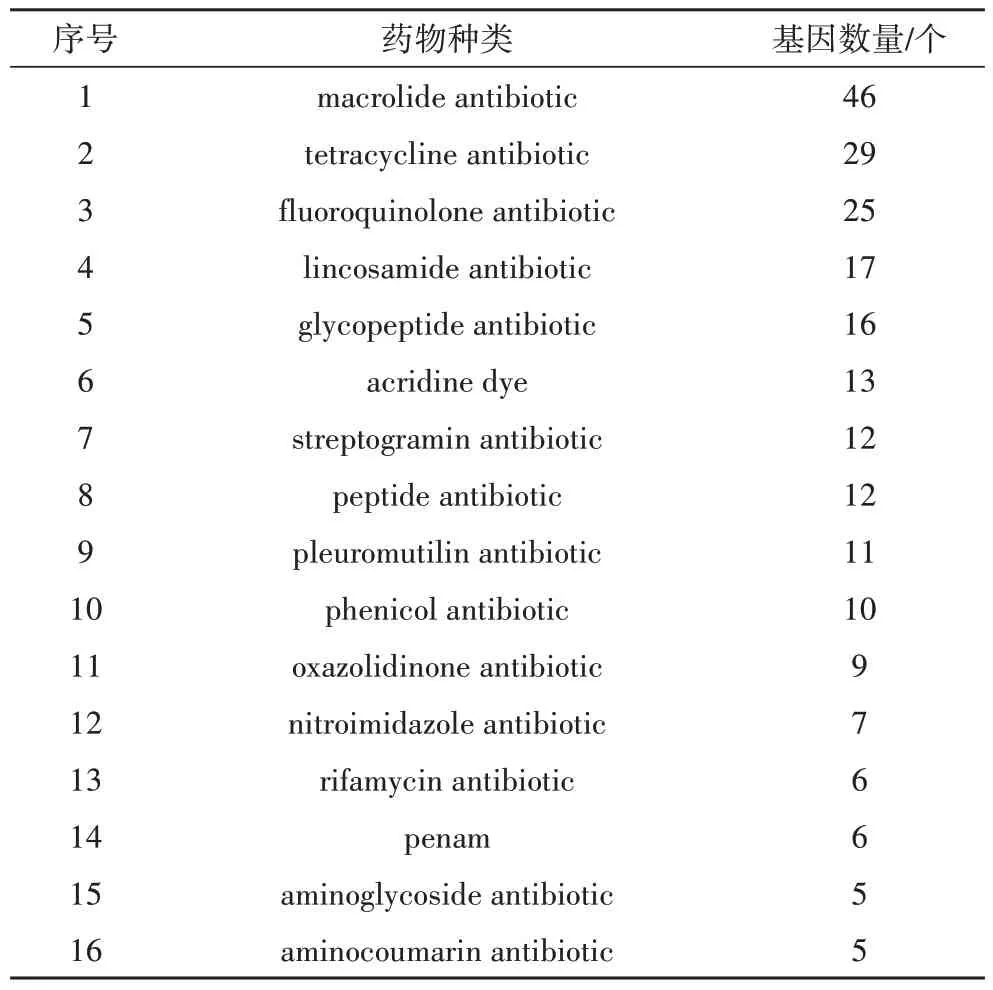
表2 无乳链球菌S.agalactiae CS2108耐药基因统计表
3 讨论与结论
分子生物学技术的飞速发展及大量罗非鱼无乳链球菌基因组序列的公布,促进了对无乳链球菌基因组特征、流行病学、病原学、检测方法和防控措施等方面的研究,为有效预防和治理提供了理论依据。
无乳链球菌基因组平均大小在2.05 Mb 左右,S.agalactiaeCS2108 的基因组大小为2.19 Mb,略大于平均值。染色体基因编码2 104个蛋白质,小的质粒编码6 个蛋白质。相较于其他病原微生物的基因组和编码的蛋白质数量都较少,其他基因序列的功能还需要进一步研究。基因组岛是有横向起源迹象的一部分基因组,每个基因组岛与多种生物功能相关、与共生或病原机理相关、与生物体的适应性相关,S.agalactiaeCS2108 的基因组中含有8 个基因组岛,包含基因数量在10~63 个不等,每个基因组岛含有功能相似的基因;前噬菌体序列上往往存在一些功能基因(抗生素基因、毒力基因)增强细菌对环境的适应性或使细菌成为致病菌,S.agalactiaeCS2108 中包含2 个前噬菌体,使得成为溶源菌;CRISPR/Cas 系统是一种原核生物的免疫系统,用来抵抗外源遗传物质的入侵,比如噬菌体病毒和外源质粒。它可以识别出外源DNA、沉默外源基因的表达,S.agalactiaeCS2108 中包含6 个CRISPR/Cas,在抵抗外来病毒或者DNA入侵过程中起着重要作用。
有1 820 个基因注释到COG 功能注释中,其中约25%(462 个)的基因功能是未知的,这表明在无乳链球菌-罗非鱼互作中仍有较多的新基因功能有待深入研究;其他基因中与复制、重组、修复相关的占比最高,在宿主中病原菌要面临宿主细胞的攻击和吞噬,所以复杂的重组和修复系统非常重要;其次是与碳水化合物转运和代谢相关的基因,因为碳水化合物能为病原菌提供碳源,保证病原菌在宿主中能够存活下来[19]。
通过比较基因组、蛋白质组学和组织病理学等方法来研究强弱毒株的毒力差异,揭示了强毒株在罗非鱼体内的定植和组织损伤规律[19]。影响无乳链球菌毒力强度的基因包括荚膜多糖、表面蛋白、溶血素、CAMP 因子、丝氨酸蛋白酶和透明质酸等相关基因[20-21],但对毒力基因缺乏深入的研究,我们基因组测序中发现了20个与毒力相关的基因。
抗生素是防治鱼类疾病的主要手段,随着抗生素使用量的增多产生了越来越多的耐药性,抗生素的耐药性成为十分严峻的问题,不同的无乳链球菌对抗生素的敏感性不同。张新艳等从广东分离的无乳链球菌的耐药性相对较弱,对氨苄西林等28 种抗生素敏感,对复方新诺明等15 种抗生素不敏感[22];霍欢欢等发现海南分离到的无乳链球菌耐药性很强,大部分菌株对氨基糖苷类和喹诺酮类耐药[23]。了解无乳链球菌的耐药规律,对于指导合理使用抗菌药、防止多重耐药菌株流行和扩散、保障水产品质量安全具有重要意义。
无乳链球菌为了适应不同环境下宿主体内的生活环境,会朝着不同的方向变异,同时基因组也会发生相应的变化。环境因素在无乳链球菌致病过程中有促进作用,例如高温、高亚硝酸盐、高氨氮、低溶解氧及pH 值等[24]。无乳链球菌通过躲避吞噬细胞的免疫防御,部分细菌被黏附吞噬后可在巨噬细胞内存活、生长和繁殖,随巨噬细胞在体内游走、侵染其他组织,还可透过血-脑屏障侵入脑组织,导致罗非鱼呈现典型的临床症状和组织病理学变化[25]。
病原、宿主与环境之间的平衡关系决定疾病发生与否,了解链球菌感染与传播途径,可以更深入地认识罗非鱼、链球菌和环境之间的关系[12]。罗非鱼摄食了携带无乳链球菌的食物进入胃肠组织后,以单个个体、成组或成链的形式附着在肠细胞的顶端边缘,穿越上皮细胞,在胃肠道黏膜上富集和增殖,然后随体液循环进入各组织内,最后突破血脑屏障,进入脑部,导致脑膜炎,破坏罗非鱼的脑神经,进而出现转圈圈的现象[25]。在实际的养殖环境下,高浓度的菌液环境难以实现,链球菌通过口腔进入罗非鱼的胃和肠道,可能是自然条件下感染罗非鱼的主要传播途径。对S.agalactiaeCS2108 菌株进行全基因组测序分析,将为罗非鱼无乳链球菌的预防和防治提供理论支持。
猜你喜欢
云南化工(2021年6期)2021-12-21
现代畜牧科技(2021年3期)2021-07-21
农药科学与管理(2019年6期)2019-11-23
当代水产(2019年4期)2019-05-16
当代水产(2018年8期)2018-11-02
农家之友(2018年12期)2018-03-12
华南农业大学学报(2015年5期)2015-12-04
医学研究杂志(2015年8期)2015-06-22
当代畜禽养殖业(2014年11期)2014-02-27
当代畜禽养殖业(2014年1期)2014-02-27
