
Fast-tracking drug development with biomarkers and companion diagnostics
2024-05-17 07:04NoreenMcBreartyDevikaBahalSusoPlatero
Noreen McBrearty, Devika Bahal, Suso Platero
Discovery Life Sciences, Newtown, PA 18940, USA.
Abstract To fully implement precision medicine, a deeper understanding of biomarkers, companion diagnostics, and their use in clinical trials is needed.Here, we describe key events in biomarker discovery and clinical trial design, and how those stages may be streamlined to fast-track approval of companion diagnostics (CDx).We discuss crucial qualities of a successful CDx that include understanding the prevalence of the marker in the intention to treat population, careful consideration of the scoring scheme that will be used in later clinical trial stages, and reliability of the performance of the CDx, in addition to other necessary features.
Keywords: Biomarkers, companion diagnostics, oncology, personalized medicine, drug development, clinical trials,regulatory approval
INTRODUCTION
The power of biomarkers has been understood for decades, but their utility in personalized medicine is only beginning to be realized through their use as companion diagnostics (CDx).The Food and Drug Administration (FDA) defines CDx as a “medical device, often an in vitro diagnostic (IVD), which provides information that is essential for the safe and effective use of a corresponding drug or biological product”(https://www.fda.gov/medical-devices/in-vitro-diagnostics/companion-diagnostics).Biomarkers, present in the targeted population, can provide critical information that informs patient treatment decisions.The first companion diagnostic was approved over thirty years ago, using the biomarker Her2 to select patients likely to benefit from treatment with trastuzumab.The more recent blockbuster approval of the first CDx used alongside an immunotherapy, the PD-L1 IHC 22C3 pharmDx test for pembrolizumab, highlights the utility of biomarkers as companion diagnostics, their ability to inform treatment decisions and improve patient outcomes.Though the benefits of companion diagnostics are acknowledged and far-reaching, translating them from preclinical research into the clinic has proven challenging.As of 2023, there are approximately 170 Food and Drug Administration (FDA) cleared or approved CDx[1], including imaging and device companion diagnostics.However, most CDx target the same few markers, with only thirty-four unique markers approved.For example, there are thirty-two CDx targeting the biomarker EGFR and twenty-five targeting Her2[1], some of which are the same test approved for use with different therapies.Challenges to approval considered herein include poor preclinical models, lack of prevalence in the patient population,poor assay development, regulatory hurdles, and inadequate clinical trial design.We discuss each phase of biomarker and companion diagnostic development, beginning with the early research stage, through the clinic, and ultimately to FDA approval [Figure 1].Furthermore, we discuss the future of the field, including the tremendous growth potential and improvements in the regulatory process that aim to expand the approval of CDx so that more patients may benefit.
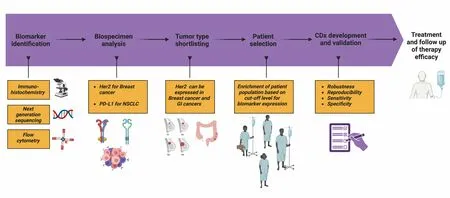
Figure 1.Ideal course of CDx Development includes initial steps of biomarker identification using varied modalities, analysis of biospecimens for biomarker level quantification leading to cancer indication finalization based on biomarker expression.These initial steps inform patient selection, companion diagnostic development and validation, and eventual large-scale treatment and follow-up for treatment efficacy.
PRECLINICAL TESTING: IMPORTANCE OF APPROPRIATE MODELS AND IDENTIFYING BIOMARKER PREVALENCE IN THE INTENTION-TO-TREAT POPULATION
Cancer is known to be a complex disease in which the same cancer type can behave differently in different populations.The consequence of these individual differences is that within a population of patients suffering from the same cancer, the mainstay therapy fails to achieve efficacy in many patients.This is where the identification of a biomarker that selects the responders for treatment is of utmost importance.This type of biomarker is known as a predictive biomarker and its identification often begins at the first stage of drug development, that is, the research phase.One of the best examples of this type of biomarker was seen when BRCA1 and BRCA2 mutations in a cell line made the cells susceptible to PARP inhibitors[2].In this instance, the BRCA mutations were predictive biomarkers for treatment efficacy in a subset of breast cancers[3].A similar approach was used when classifying the BRAF gene mutation as a predictive biomarker in melanoma tumors[4].Studies confirmed that treatment with a BRAF inhibitor successfully reduced cancer cell proliferation in the presence of the BRAF mutation[4], while other drugs, such as EGFR inhibitors, were found to be ineffective[5].Thus, screening for predictive biomarkers enables testing drugs in the right population, and ensures that patients receive the most effective treatments while also avoiding therapy to which their tumors are unlikely to respond.
The FDA classifies a biomarker as “a defined characteristic that’s measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions”[6].Biomarkers are the pillars on which companion diagnostics are built.A companion diagnostic is an in vitro test that provides information that ensures that the therapeutic product to be used will be effective and safe[7].Hence, for a biomarker to qualify as a companion diagnostic, it needs to be expressed in a particular tumor population, play a key role in the maintenance or progression of cancer, have high reproducibility in a homogenous population of patients suffering from the same subtype of the tumor, and have proof, through clinical trials, that manipulating the target will bring about an appreciable, positive change in the disease outcome[8].In addition, for a biomarker to be used effectively as a CDx, it needs to be reproducible and reliable in a clinical trial setting.That is, when anyone runs the test anywhere, they will get the same results every time the test is performed.
One of the major drawbacks of preclinical experiments is that the tumor cell lines studied in vitro, and model organisms studied in vivo, are not always representative of the cancer subtypes seen in patients.Common model organisms used in drug discovery and development include mouse, zebrafish, yeast, andC.elegans;however, they often fail to fully recapitulate the complex biology observed in humans.The lack of relevancy in some tumor models is a major contributor to the inability to translate basic research findings into the clinic, underscoring the importance of confirming that preclinical tumor models accurately reflect the biology driving tumor growth in the intention-to-treat patient population.
The paradigm-shifting approach of precision medicine is to understand the molecular drivers of the patient’s disease and target them accordingly.For example, in colorectal cancer (CRC) patients, a large European study found that RAS mutations affect 43%-44% of CRC[9]patients, whereas BRAF mutations were less common, affecting only 8%-12% of CRC patients[10].This difference in driver mutations dictates different treatment courses within the same tumor type.BRAF mutations are much more common in melanoma patients, with approximately 40% containing a mutation in this gene[11].Of those with a BRAF mutation, a Phase III study performed in melanoma patients found an objective response rate of 70% after combined therapy with BRAF/MEK inhibitors[12].This finding highlights the increased efficacy that a companion diagnostic can provide by screening for patients who harbor a biomarker indicative of treatment response, rather than treating cancers of the same subtype in the same manner and disregarding the genomic profile driving the cancer.
A successful method to assess biomarker prevalence in cancer indications of interest is performing a large tumor screen using archived or fresh patient tissue.This method has been shown to help determine the indication with the highest expression level of the marker and inform clinical trial design.An example of the importance of identifying biomarkers was demonstrated in clinical trials and observational studies that showed patients with KRAS mutations who were given anti-EGFR treatment did not derive benefit from it[13], significantly reducing the overall response rate of the drug.By knowing the subset of patients that express a particular biomarker, we can stratify the patient population to those that are likely to respond to therapy while also eliminating unnecessary treatment in those unlikely to respond and guide them to a more appropriate and efficacious treatment option.In large part, the regulatory approval of the CDx follows the contemporary drug approval process[Figure 2], wherein there is a preclinical phase followed by clinical trials and approval.To initiate a clinical trial of a novel drug, an Investigational New Drug (IND) application must be granted by the FDA unless the drug meets IND exemption criteria.If a CDx will be used to assess safety and effectiveness data, an investigational device exemption (IDE) must be obtained prior to Phase I.This will allow its use to assess clinical trial results.To evaluate the safety and effectiveness of the CDx itself, the regulatory submission process may occur through premarket notification submission [510(k)] or premarket approval application(PMA), depending on the assessed risk and ability to mitigate risk.Most CDx are classified as Class III medical devices which require PMA; however, some may be classified as Class II medical devices which only require 510(k) submission.If the CDx is already approved for use with another drug or a different cancer indication, the process of the approval of the new therapy is quicker since an IDE may not be required.As per the FDA guidelines, CDx are reviewed primarily by the Center for Devices and Radiological Health (CDRH) and drug approvals are done by the Center for Drug Evaluation and Research(CDER) and Center for Biologics Evaluation and Research (CBER), depending on if the drug is a chemical substance or a biological one; hence, both branches of the FDA should be approached simultaneously if a CDx is evaluated with a new drug.
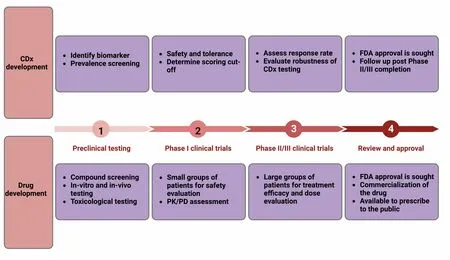
Figure 2.Co-development of CDx with therapy.
A significant consideration to designing the proper clinical trial should be given, as a poorly planned trial can sabotage an otherwise successful drug.The length of time it takes to enroll patients expressing a specific mutation or amplification targeted by treatment can delay drug approval.In addition to using biomarkers to select patients likely to respond, N-of-1 trials may speed up the drug development process by allowing a single patient to be the sole point of analysis.This model may be most helpful in clinical trials that have difficulty recruiting patients, as can be observed with rare diseases and personalized medicine.While this model poses challenges in oncology, particularly in patients with metastatic disease, personalized trials enable a patient-centric focus where patients act as their own controls and undergo intra-patient dose escalation to determine the most appropriate intervention.Further, repurposing previously approved drugs in novel combinations is an ongoing area of research due to the potential to reduce approval timelines and cost, and improve response rates.Since the discovery programs have occurred and the safety, dosing, and pharmacokinetic profile of each drug are known, the therapy can move more quickly into trial[14,15], where the safety profile in combination can be studied.This synergistic approach may streamline the regulatory process and seek to attack cancer through multiple, non-redundant mechanisms[15]to bring more effective therapies to the clinic more quickly.
PHASE I CLINICAL TESTING: LOCKING IN THE CDX
Currently, the primary goal of a Phase I clinical trial is to identify not only the safety and tolerability, but also the dose of the experimental drug.If the experimental drug is found to be effective in a subset of patients expressing the target, a companion diagnostic may be initiated to fast-forward the drug development.While the FDA does not always require the simultaneous approval of a drug and its diagnostic, co-development is deemed essential when the CDx is used for selection of the patient population.If the CDx has been found to be required for the safe and effective use of the drug, the FDA will most likely not approve the therapy or new therapeutic indication unless, and until, the CDx has been granted approval.The HercepTest, an IHC assay detecting Her2, was the first FDA-approved companion diagnostic, obtaining approval in 1998.The HercepTest defined the model for the co-development of a drug alongside its companion diagnostic and successfully identified responsive patients in several breast and gastric cancer clinical trials.Since its approval more than 25 years ago, HercepTest continues to demonstrate its clinical significance, facilitating the approval of Herceptin for early-stage and metastatic Her2+ breast cancer and metastatic Her2+ gastric cancer.Despite the early misconception that personalized medicine may prove financially restrictive, Herceptin remains in the top 5 highest-grossing oncology drugs.Other well-known examples of approved companion diagnostics include the PD-L1 CDx for Keytruda, c-KIT CDx for Gleevec, and TP53 CDx for Venclexta.
The important points of consideration during the development of a companion diagnostic at the Phase I stage are to confirm that the test gives suitable, sensitive, robust, and reproducible signals.The key to establishing a suitable signal lies in being able to distinguish a dynamic range of expression, meaning that the test can differentiate between negative, low/moderate, and high expression, as this criterion may be used to form the critical scoring cut-off that determines if a patient enters treatment [Figure 3].While simply the presence or absence of some biomarkers inform treatment response, such as many immune markers, the relative intensity of expression is important for others, including Her2 and other tumor markers.For Her2 testing in breast cancer, a four-tier criteria system (i.e., 0, 1+, 2+, 3+) is used to understand the expression.If Her2 expression is considered 0 or 1+ based on the CDx IHC test, the tumor is deemed Her2-negative.A score of 0 indicates that no staining or incomplete membrane staining that is barely perceptible is observed in less than 10% of invasive tumor cells, while a 1+ indicates that faint/barely perceptible membrane staining is detected in greater than 10% of invasive tumor cells[16].Such tumors have been found mostly to be nonresponsive to Herceptin; thus, Her2-negative patients will not receive Herceptin treatment but instead will be guided towards a different therapy, which will likely achieve a better response.If Her2 expression is considered 3+ based on the CDx test, defined as complete circumferential membrane staining in greater than 10% of invasive tumor cells, the tumor is considered Her2-positive and treatment with Herceptin is indicated.Tumors that score 2+, defined as incomplete and/or weak to moderate membrane staining in greater than 10% of invasive tumor cells, are considered equivocal and require further fluorescencein situhybridization (FISH) testing in order to confirm amplification and indicate the course of treatment[17].
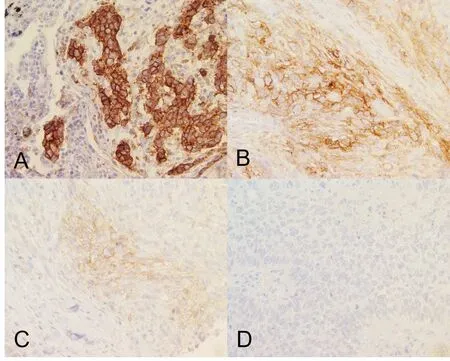
Figure 3.Tumor cell staining showing (A) high, (B) moderate, (C) low, and (D) negative PD-L1 membrane expression.Images taken at 40x magnification.As per FDA guidelines for Keytruda eligibility, tumors shown in (A-C) will be eligible for anti-PDL1 therapy.
Robustness refers to the ability of the test to show clinical relevance and identify patient populations most likely to derive benefit from the drug while eliminating those unlikely to respond and suffer unnecessary side effects.An intentional consequence of this approach is that a robust companion diagnostic should restrict enrollment to the most likely responders, stratifying the patient population to those who express the marker targeted by the drug.
Reproducibility describes the ability of the test to be performed by various trained operators in different qualified labs and still produce the same result.This is critical as without it, the lab or technician performing the test may influence the outcome.These parameters must be put in place and the companion diagnostic defined during the Phase I trial so that a validated CDx may be used in subsequent trial phases.
Lastly, an ill-chosen scoring scheme and cut-off can severely affect the outcome of a clinical trial, leading to failure and rejection of approval.This is best demonstrated by the CheckMate-026 clinical trial evaluating the efficacy of Opdivo as a first-line immunotherapy in the treatment of non-small cell lung carcinoma(NSCLC) patientsversusthe Keynote-010 study, which evaluated Keytruda in this indication.The Keynote-010 study was able to correlate PD-L1 expression ≥ 50% with response to treatment, whereas the CheckMate-026 used a different biomarker assay for PD-L1 along with a lower cut-off of ≥ 5%, which is widely considered to have contributed to its failure to meet the primary endpoints[18].The financial consequences of this failure were a nearly 20% decline in Bristol-Myers Squibb (BMS) stock price and a class action lawsuit brought against the company[19].Interestingly, later clinical trials have lowered the > 50% cutoff for Keytruda, but it still has maintained its dominance over Opdivo.
Importantly, evaluation of the cut-off and scoring scheme in each indication is essential.For example, Her2 testing in gastric cancer varies from that in breast cancer due to differences in tumor biology, heterogeneity of the marker, and incomplete membrane staining more commonly observed in gastric cancer[20].Given the differences observed in Her2 expression, the College of American Pathologists (CAP) and FDA have adopted appropriate, individual scoring systems for breast and gastric indications, so as not to unnecessarily reject potential responders from receiving treatment[20].
PHASE II/III CLINICAL TESTING: CONFIRMING CDX EFFICACY AND VERSATILITY
Following appropriate IDE approval, the CDx is then authorized to be used in Phase II/III clinical trials with its associated therapy at the pre-approved cut-off score that was determined based on the preclinical and Phase I trial data.During the Phase II trial, an important consideration for the success of the CDx is rigorously analyzing the cut-off.This is an integral step since, at this stage in the CDx development,treatment responses based on the stratification of patients are obtained.For example, in a recent study that analyzed the response of PD-L1 inhibitor Tecentriq based on the tumor mutational burdens (TMB) of different cancers, the initial cut-off value was TMB-H 10 mut/Mb[21].This cut-off value was based on Keytruda and has been approved by the FDA for patients expressing TMB-H 10 mut/Mb[22,23].Interestingly,Tecentriq was found to be effective in patients who had TMB-H 16 mut/Mb and was significantly higher than in patients who had TMB-H 10 mut/Mb and 16 mut/Mb[21].Herein, we see that in a direct comparison between the two drugs, Keytruda had an objective response of 29.0% in their TMB-H 10 mut/Mb group[23],while Tecentriq had an objective response of 38.1% in their TMB-H 16 mut/Mb group[21], which would be obscured had the researchers not further stratified in the Tecentriq study[21].
Another key advantage of having a CDx developed for a drug is that, in the future, it can be extended to testing and eventual approval in similar classes of drugs from different companies.A case in point is the FoundationOneCDx, which was used in both the Tecentriq[21]and the Keytruda[23]studies.Since these drugs are PD-L1 inhibitors, testing is more streamlined when the CDx has already been approved for the same class of drug.The fast-tracked approval of Osimertinib, currently the only FDA- and EMA-approved thirdgeneration EGFR tyrosine kinase inhibitor (TKI), is one of the most striking examples of using a previously approved CDx to facilitate and expedite approval of a new drug[24].Osimertinib is approved for first-line treatment of EGFR T790M mutation-positive metastatic NSCLC as detected by an FDA-approved test.The drug discovery program responsible for Osimertinib was initiated in 2009 and the drug was ready to be evaluated in clinical trials beginning in 2013.The anti-tumor effect of the drug was so significant, with activity observed in the first cohort of patients treated[25], and in 2015, the FDA granted accelerated approval for the treatment of metastatic EGFR T790M mutation-positive NSCLC[26].The timeline from discovery to approval for Osimertinib was six years, compared to the average approval time of 10-15 years from discovery to drug approval[27], a journey undoubtedly streamlined by the existence of an appropriate and approved CDx.
This is also reflected in the approval of Herceptin, Perjeta, and Kadcyla by the FDA, which are all used with the HercepTest CDx[1].Moreover, the same CDx can be developed to stratify patients to treat different cancers with the same drug.For example, Keytruda and its CDx are FDA-approved for the treatment of NSCLC, Cervical Cancer, Head and Neck Squamous Cell Carcinoma (HNSCC), Esophageal Squamous Cell Carcinoma (ESCC), and Triple-Negative Breast Cancer (TNBC).Here, we see that the development and validation of a single CDx for one targeted therapy against a tumor type can, in due course, be applied as a therapy for multiple, diverse tumor types.This method is cost-effective and can increase revenue streams for the participating companies in an extremely cost-prohibitive drug development system.Furthermore, it uses less tissue for testing, which is an advantage in tumors with a small amount of sample.The subsequent fast turnaround time for the approval of the therapy and its CDx for the treatment of different cancers is valuable not only for pharmaceutical and medical device entities but also for patients who have quicker access to better therapeutic drugs.
Currently, tissue-based biopsy tests make up the majority of approved CDx, but liquid biopsy tests analyzing circulating tumor DNA (ctDNA) are emerging as important tools in precision medicine.The first FDA-approved ctDNA-based CDx was the Cobas EGFR Mutation Test V2, approved in 2016.Since then,only three more ctDNA-based CDx have been approved by the FDA[28].These CDx inform the management and treatment of metastatic disease and can be used to identify patients at the highest risk of recurrent disease since residual ctDNA after local therapy is indicative of molecular residual disease (MRD)[28].The potential of ctDNA as a prognostic biomarker and utility in patient enrichment for clinical trials is apparent;however, these assays have proven challenging to clinically validate and are not yet sufficient for use as an early endpoint to support drug approvals[28].While there has been discordance noted in the ctDNA tests compared to traditional biopsies, including a higher rate of false negatives due to low sensitivity[28], there is no doubt that the many advantages of liquid biopsies over traditional biopsies support their continued development and improvement for use in CDx.Despite the focus on ctDNA in clinical trials, the FDA only recently released guidance for ctDNA in early-stage solid tumor drug development, designed to help sponsors use ctDNA as a biomarker in early-stage clinical trials[29].Further standardization and guidance have been released by the BloodPac consortium, a not-for-profit group consisting of industry and academic partners, which describes guidelines to be used in analytical validation protocols and standardized methods for ctDNA assays[30].Both efforts seek to bridge some of the gaps that have so far prevented establishing the clinical utility of ctDNA.Given the overall promise of ctDNA-based CDx, it is imperative that regulatory bodies focus efforts on keeping up with the technology and facilitating its use in clinical trials.
While every step of the CDx development ideally occurs in tandem with the clinical trial of its corresponding drug, there can be instances when the need for the CDx arises late in the drug development process.In such cases, there can be a provision granted by the FDA for conditional approval of the CDx to be tested directly in the Phase III trial of the drug, while the CDx also goes through the final stages of its development.Keytruda was given approval by the FDA in 2017 for the treatment of patients with solid tumors of Microsatellite Instability High (MSI-H) status[31].Many years later, the FoundationOneCDx for identifying MSI-H status solid tumors was approved by the FDA[32]to aid in identifying patients who would respond to this treatment.While Foundation Medicine did not take the regular route for the validation of their CDx in this scenario, the existing approval for Keytruda as a treatment in the cohort of patients that had MSI-H solid tumors allowed the CDx assessment to be incorporated in ongoing clinical studies in 2020[33]and led to its approval in 2022[32].A similar scenario was also seen with Vitrakvi, wherein the drug was given the approval for use in solid tumors with NTRK gene fusions[34,35]and the CDx was consequently approved after testing in Phase I and II clinical trials[36].
At the end of the Phase II/III trial, a PMA CDx will be analyzed for its clinical utility and label considerations[37].Once approved by the FDA, the drug moves into the market launch and commercialization phase.While the road to FDA approval is rigorous, the post-approval phase can be even more tumultuous[38].In the development of Xalkori against NSCLC, drug developer Pfizer and CDx developer Abbott Molecular Inc.were initially reported to have difficulty agreeing on the best approach to CDx development, but the path to approval became more defined once they viewed one another as collaborators[39].The need for thoughtful partnering cannot be overstated since work from development to clinical trials to post-approval marketing and beyond requires a team effort among multiple players.Basic research organizations, pharmaceutical companies, CROs, hospitals, and medical device companies are all indispensable contributors that need to work seamlessly to bring patients the state-of-the-art care they deserve.
CONCLUSION
While the first companion diagnostic was approved as early as 1998[40], the field was slow to take off but currently shows promising growth[41].Given the power of CDx in the diagnosis and treatment of diseases,the FDA is making strides to improve the approval process.In 2014, the FDA released its final regulatory guidance on CDx development that seeks to assist sponsors through the complex approval process.In line with the increased focus on CDx, the global CDx market was valued at $2.43 billion in 2019 and is forecasted to grow at a rate of 18.9% to an estimated $9.72 billion in 2027[41].While the market for CDx continues to expand, many challenges must be addressed to fuel the growth of newer drugs and their CDx.The FDA recently released guidance describing how sponsors can expand the development and labeling of CDx so that it no longer applies to a single drug but to a specific group of oncology therapeutic products[42].Evidence of this broadening in CDx approval came in 2022 when the FDA approved the FoundationOneCDx to determine which NSCLC patients, whose tumors harbor EGFR exon 19 deletions or exon 21 substitutions, are likely to benefit from EGFR tyrosine kinase inhibitors (TKIs) if the drug has previously been approved in that indication, demonstrating a willingness to adjust requirements to allow faster CDx development and expanded application.
Lastly, the importance of identifying a robust biomarker and developing tests that can detect it with accuracy and reproducibility is key to fast-tracking the clinical development of a drug.Incorporating the best cut-offs of the biomarkers in clinical trials will facilitate superior responses from the target population and can be the deciding factor between a failed trial or a new revolutionary treatment option.Expanding the approval of indications through continued research into other cancer indications, and/or other drugs from the same family, will ensure that the scope of use of the CDx increases and more patients benefit from this advancement.The flexibility shown by the FDA in treating each CDx case as an individual[43]and quicker approval time should usher the field into never-before-seen growth for the industry and the patients who benefit from advancement in science.Fortunately, the innovative potential of CDx has united industry,academic, and regulatory bodies in the effort to personalize treatment therapies to reap the greatest benefits for current and future patients.
DECLARATIONS
Acknowledgments
The authors would like to thank Karen Kirchner for editorial assistance.
Authors’ contributions
Conceptualized, designed, drafted, and revised the manuscript: McBrearty N, Bahal D
Conceptualized, designed, and drafted the manuscript: Platero S
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
 Journal of Cancer Metastasis and Treatment2024年1期
Journal of Cancer Metastasis and Treatment2024年1期
- Journal of Cancer Metastasis and Treatment的其它文章
- Feature interview with Dr.William C.CHO -“Clarivate 2023 Highly Cited Researcher”
- Prognostic value of clonal evolution identified by sequential FISH in untreated chronic lymphocytic leukaemia
- Mechanical force-mediated interactions between cancer cells and fibroblasts and their role in the progression of hepatocellular carcinoma
- Leveraging metformin to combat hepatocellular carcinoma: its therapeutic promise against hepatitis viral infections
- Regulation and function of the RSK family in colorectal cancer