
藤黄酸和雷帕霉素共载脂质体的制备及协同抗肿瘤活性研究
2024-05-14 10:10丁欢欢姜先梅刘红梅涂柯蓉蔡璐璐周先礼
中草药 2024年9期
丁欢欢,姜先梅,刘红梅,齐 瑶,涂柯蓉,陈 琳,蔡璐璐*,周先礼*
·药剂与工艺·
藤黄酸和雷帕霉素共载脂质体的制备及协同抗肿瘤活性研究
丁欢欢1, 2,姜先梅2, 3,刘红梅2, 3,齐 瑶2, 3,涂柯蓉2, 3,陈 琳1,蔡璐璐2, 3*,周先礼1*
1. 西南交通大学生命科学与工程学院,四川 成都 610031 2. 四川省医学科学院·四川省人民医院,药学部,四川 成都 610072 3. 电子科技大学医学院,个体化药物治疗四川省重点实验室,四川 成都 610072
制备共载藤黄酸和雷帕霉素的脂质体(liposomes co-loaded with gambogic acid and rapamycin,GR@Lip)并优化其处方,研究GR@Lip的体外抗肿瘤机制、体内药动学和生物分布。以包封率、载药量及粒径为评价指标,通过单因素和正交设计实验筛选GR@Lip的最佳处方,并对其进行表征和稳定性研究;通过CCK-8法和流式细胞术考察GR@Lip对肿瘤细胞增殖和凋亡的影响,细胞划痕实验与Transwell实验考察GR@Lip对肿瘤细胞迁移与侵袭的影响,透射电子显微镜(transmission electron microscope,TEM)和免疫荧光考察GR@Lip对肿瘤细胞自噬的影响,液相色谱-质谱联用技术(liquid chromotography with mass spectrometry,LC-MS)和活体成像仪研究GR@Lip的体内药动学和生物分布。GR@Lip的最佳处方为制备温度40 ℃,药脂比1∶1∶20,磷脂与胆固醇比例4∶1,超声功率195 W,超声时间5 min,水化介质为超纯水,水相pH值为7.1;该方法制备的GR@Lip藤黄酸包封率为(97.27±2.76)%,雷帕霉素包封率为(96.58±3.82)%,藤黄酸载药量为(3.29±0.44)%,雷帕霉素载药量为(4.91±0.44)%。TEM形态观察显示GR@Lip呈球形,动态光散射(dynamic light scattering,DLS)检测其平均粒径为(157.19±1.74)nm、ζ电位为(−22.1±1.3)mV,且具有良好的稳定性。体外抗肿瘤活性实验结果显示,藤黄酸和雷帕霉素联用能协同抑制肿瘤细胞增殖、迁移和侵袭,并显著促进肿瘤细胞凋亡,增强自噬。此外,体内药动学和生物分布结果显示,GR@Lip可在体内滞留更长时间且具有良好的肿瘤靶向性。成功制备了GR@Lip,揭示其具有通过多途径协同抗肿瘤的活性,并显著改善了藤黄酸和雷帕霉素的药动学行为,为进一步体内研究和未来临床应用提供了实验依据。
藤黄酸;雷帕霉素;共载脂质体;抗肿瘤;联合用药;体内药动学;生物分布;增殖;凋亡;迁移;浸袭;自噬
恶性肿瘤是全球范围的重大公共卫生问题,严重威胁人类的生命健康。化疗作为治疗恶性肿瘤的重要手段之一,为肿瘤患者的生命健康做出了巨大的贡献。雷帕霉素是雷帕霉素靶蛋白(mTOR)的天然抑制剂,已被FDA批准用于治疗肾癌,其衍生物已用于脑肿瘤、胰腺癌和乳腺癌等的治疗。由于肿瘤病理机制复杂多元,仅依靠单一抗肿瘤机制的药物治疗很难获益。因此,探寻创新型高效低毒的药物联用模式已成为肿瘤治疗的有效策略。中药成分具有多靶点、多环节、多途径的特点和较低的不良反应,与化疗药物联用可减轻临床症状、降低不良反应、防止耐药和复发[1],在肿瘤治疗中具有巨大前景。藤黄酸是从中药藤黄中提取的一种天然化合物,可有效抑制多种肿瘤生长。藤黄酸也被证实可与化疗药物如多柔比星[2]、吉西他滨[3]等联用,协同增强抗肿瘤疗效[4]。因此,本研究旨在探索藤黄酸与雷帕霉素的组合策略是否可以改善肿瘤治疗效果,并初步研究其协同抗癌机制。
然而,藤黄酸具有水溶性差、刺激性强且选择性较低的缺点,雷帕霉素难溶于水且单用易产生耐药性导致临床应用受到严重阻碍,基于纳米技术的药物共递送系统在解决上述缺陷方面表现出显著的优势[5]。研究显示,多药共载纳米制剂可在保持药物活性的同时显著增强疗效[6]。脂质体作为目前研发比较成熟的装载疏水性药物的制剂,其磷脂双分子层结构可以更有效地被细胞摄取,从而提高药物递送效能[7-8]。此外,基于脂质体的被动靶向作用,可以改善药物在体内的药动学行为,增强肿瘤靶向性。本实验将藤黄酸和雷帕霉素共载于脂质体中,通过单因素和正交设计试验对处方工艺进行优化,研究其对多种肿瘤细胞增殖、凋亡、迁移和侵袭以及自噬的影响,并进一步研究其体内药动学和生物分布,初步探索双药共载脂质体的抗肿瘤作用机制,以期为藤黄酸临床制剂的应用转化提供依据。
1 仪器与材料
1.1 仪器
XSE205型分析天平,梅特勒托利多仪器有限公司;RE-2010型旋转蒸发仪,成都康宇科技有限公司;SHB-III型循环水式多用真空泵,郑州长城科工贸有限公司;WB-2000型水浴锅,郑州长城科工贸有限公司;SM-650A型超声波细胞粉碎机,南京舜玛仪器设备有限公司;Litesizer 500型纳米粒径及Zeta电位分析仪,安东帕(上海)商贸有限公司;HT7820型低电压透射电子显微镜(TEM),日本日立公司;Agilent 1260型高效液相色谱系统,美国安捷伦公司;色谱柱Sun Fire C18(250 mm×4.6 mm,5 μm),美国Waters公司;SB25-12DTD型超声波清洗器,宁波新芝生物科技股份有限公司;i-Pure Pro2智能型纯水/超纯水机,杭州泽南科技有限公司;FD-1C-50型冷冻干燥机,北京博医康实验仪器有限公司;75004240型离心机,百乐科技有限公司;Herocell 240型二氧化碳培养箱,上海润度生物科技有限公司;Epoch2型酶标仪,美国伯腾仪器有限公司;ICX41型生物显微镜,宁波舜宇仪器有限公司;NovoCyte 2070R型流式细胞仪,安捷伦科技有限公司;AniView 100型多模式动物活体成像系统,广州博鹭腾生物科技有限公司;AB Sciex Qtrap 5500型液相色谱-质谱联用仪,日本岛津公司。
1.2 材料
藤黄酸,批号20210122,质量分数98%,南京康满林化工实业有限公司;藤黄酸对照品,批号M0802AS,质量分数≥97%,大连美仑生物技术有限公司;卵磷脂(批号C14422665)、雷帕霉素(批号c12477612,质量分数98%),上海麦克林生化科技有限公司;雷帕霉素对照品,批号J17GB155257,质量分数≥99%,上海源叶生物科技有限公司;和厚朴酚对照品,批号20221027,质量分数≥98%,北京北方伟业计量技术研究院;子囊霉素对照品,批号PR230820-29,质量分数95.3%,广州亮化化工有限公司;吲哚菁绿(indocyanine green,ICG),批号22Z144-D1,质量分数90%,上海甄准生物科技有限公司;胆固醇,批号B80859,艾伟拓(上海)医药科技有限公司;无水乙醇,分析纯,成都金山化学试剂有限公司;甲醇,分析纯,成都市科隆化学品有限公司;乙腈,色谱纯,Sigma-Aldrich(上海)贸易有限公司;冰乙酸,色谱纯,天津市科密欧化学试剂有限公司;RPMI 1640、DMEM培养基,批号812338、8122762,赛默飞世尔生物化学制品有限公司;胎牛血清、胰蛋白酶,批号H20051277、SS1059,美国Gibco公司;CCK-8(批号CR2310019)、磷酸盐缓冲液(PBS,批号23314817),武汉赛维尔生物科技有限公司。
小鼠胰腺癌Pan02细胞和小鼠宫颈癌U14细胞均购自武汉华尔纳生物科技有限公司,批号分别为SAc0135和CTCC-400-0316;小鼠乳腺癌4T1细胞购自中国科学院典型培养物保藏委员会细胞库,批号250362。Balb/c小鼠,雌性,5~6周龄,购自斯贝福(北京)生物技术有限公司,质量合格证编号为110324241101832765。所有动物饲养在同一环境下,环境温度为(24.0±1.0)℃,相对湿度维持在55%~65%。本实验相关动物实验遵循四川省人民医院实验动物研究所有关实验动物管理和使用的规定,均符合3R原则。
2 方法与结果
2.1 藤黄酸与雷帕霉素联用比例的筛选
通过CCK8法测定藤黄酸、雷帕霉素及不同联用比例对4T1细胞的增殖毒性,将处于对数生长期的4T1细胞以5 000个/孔接种在96孔板中,孵育24 h后弃去旧培养基,加入不同质量浓度的药物继续培养24 h,然后加入CCK-8增殖检测试剂,37 ℃孵育2 h后,通过酶标仪测定450 nm处的吸光度(),并计算半数抑制浓度(half maximal inhibitory concentration,IC50,单位为μg/mL)及联合指数(combination index,CI,CI=A/IC,A+B/IC,B)。其中,A和B代表2种不同药物,IC,A和IC,B是表示A、B 2种药物单独使用使生长抑制率达时的药物质量浓度,A和B是A药和B药联合使用使生长抑制率达时2种药物的质量浓度)[9]。CI=1表示加和作用,CI<1表示协同作用,CI>1表示拮抗作用。结果见表1,当藤黄酸与雷帕霉素物质的量比为1∶1时,CI=0.772<1,协同效果最好,因此,后续选择该比例制备共载脂质体。

表1 藤黄酸与雷帕霉素联用比例筛选
2.2 GR@Lip制备方法的考察
2.2.1 薄膜水化法 精密称取藤黄酸5 mg、雷帕霉素7.28 mg、胆固醇9.23 mg与大豆卵磷脂72.43 mg(药脂比为物质的量比1∶1∶15,磷脂与胆固醇比例为物质的量比4∶1),溶于3~5 mL无水乙醇中,利用旋转蒸发仪去除溶剂,待圆底烧瓶内部形成均匀薄膜后,继续旋蒸3 h以完全除去残留溶剂。加入5 mL超纯水水化1 h,超声5 min(195 W),依次过0.45 μm及0.22 μm滤膜,即可得到GR@Lip。
2.2.2 逆向蒸发法 精密称取藤黄酸5 mg、雷帕霉素7.28 mg、胆固醇9.23 mg与大豆卵磷脂72.43 mg(药脂比为物质的量比1∶1∶15,磷脂与胆固醇比例为物质的量比4∶1),溶于3 mL氯仿中,加入1 mL超纯水,采用功率为195 W的超声波细胞粉碎机超声5 min,成乳后蒸发除去溶剂,加入5 mL超纯水水化1 h,超声5 min(195 W),依次过0.45 μm及0.22 μm滤膜,即可得到GR@Lip。
2.2.3 乙醇注入法 精密称取藤黄酸5 mg、雷帕霉素7.28 mg、胆固醇9.23 mg与大豆卵磷脂72.43 mg(药脂比为物质的量比1∶1∶15,磷脂与胆固醇比例为物质的量比4∶1),溶于3 mL无水乙醇中,40 ℃搅拌30 min。5 mL超纯水在40 ℃下搅拌30 min,将无水乙醇溶液缓慢滴入超纯水中继续搅拌30 min。旋转蒸发除去乙醇,定容至5 mL,超声5 min(195 W),依次过0.45、0.22 μm滤膜,即可得到GR@Lip。
2.2.4 考察结果 结果如表2所示,与逆向蒸发法和乙醇注入法相比,薄膜水化法制备得到的脂质体包封率最高,载药量与逆向蒸发法相近,但高于乙醇注入法,且粒径最小,表明薄膜水化法更适于制备GR@Lip,因此,后续选择薄膜水化法进一步优化其制备方法。在后续实验中通过薄膜水化法制备空白脂质体、藤黄酸脂质体(gambogic acid liposomes,GA@Lip)、雷帕霉素脂质体(rapamycin liposomes,Rap@Lip)与吲哚菁绿脂质体(indocyanine green liposomes,ICG@Lip)。

表2 制备方法考察(, n = 3)
2.3 HPLC法测定GR@Lip包封率与载药量分析方法的建立
2.3.1 雷帕霉素、藤黄酸对照品溶液的配制 精密称取雷帕霉素对照品47.72 mg,藤黄酸对照品32.60 mg,以甲醇溶解并定容于10 mL量瓶中,混匀即得雷帕霉素/藤黄酸对照品储备液,按比例稀释为所需质量浓度。
2.3.2 供试品溶液及空白脂质体对照溶液的配制 精密量取1 mL GR@Lip于量瓶中,加入适量甲醇,振荡涡旋后用甲醇定容,摇匀,0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。同法配制空白脂质体对照溶液。
2.3.3 色谱条件 色谱柱为Sun Fire C18柱(250 mm×4.6 mm,5 μm);柱温35 ℃;体积流量1.5 mL/min;检测波长288、361 nm;进样量10 µL;流动相梯度洗脱见表3,色谱图见图1。
2.3.4 系统适用性实验 取空白脂质体溶液、雷帕霉素、藤黄酸对照品溶液(雷帕霉素119.30 µg/mL、藤黄酸81.50 µg/mL)进样分析。结果表明雷帕霉素、藤黄酸与相邻组分分离度均大于1.5,拖尾因子小于1.5,对照品溶液连续5次峰面积的RSD小于2.0%,对照品溶液信噪比(/)大于10,表明方法系统适用性好。

表3 流动相梯度洗脱程序
2.3.5 线性关系考察 取系列雷帕霉素、藤黄酸对照品溶液,按“2.3.3”项下色谱条件进样测定,分别以雷帕霉素、藤黄酸的质量浓度为横坐标(),雷帕霉素、藤黄酸的峰面积为纵坐标()绘制标准曲线,得回归方程分别为雷帕霉素=14.753 4+16.543 8,2=0.999 9;藤黄酸=5.364 7+2.271 6,2=0.999 9;结果表明,雷帕霉素在14.91~357.90 μg/mL,藤黄酸在10.18~244.50 µg/mL线性关系良好。
2.3.6 精密度试验 取同一供试品溶液,连续进样6次,计算日内精密度,雷帕霉素日内检测RSD为0.13%,藤黄酸日内检测RSD为0.06%,表明方法精密度高。
2.3.7 重复性试验 精密吸取同一批GR@Lip样品6份,加适量色谱级甲醇超声溶解,制得供试品溶液,按色谱条件进样分析并平行测定3次。GR@Lip中雷帕霉素RSD为1.71%,藤黄酸RSD为1.47%,表明方法重复性良好。

图1 雷帕霉素和藤黄酸对照品(A) 及GR@Lip样品(B) 的HPLC图谱
2.3.8 加样回收率试验 精密吸取低、中、高质量浓度雷帕霉素、藤黄酸对照品储备液各5 mL,置于不同的10 mL量瓶中,再分别加入空白脂质体5 mL,并用色谱级甲醇定容,平行制备3份,再根据“2.3.2”项下所述方法配制供试品溶液。按色谱条件进样分析并平行测定3次,雷帕霉素回收率为(102.99±3.63)%,回收率RSD值为(0.68±0.34)%,藤黄酸回收率为(101.13±0.05)%,回收率RSD值为(0.73±0.28)%,表明方法准确度高。
2.4 GR@Lip包封率与载药量的测定
藤黄酸和雷帕霉素在水中的溶解度均很小(藤黄酸溶解度约为0.5 μg/mL,雷帕霉素溶解度约为0.26 µg/mL),因此,认为游离药物已在制备过程中通过微孔滤膜滤过去除。精密量取100 μL GR@Lip,另精密称取一定质量的冻干的GR@Lip,分别加入甲醇定容至1 mL进行破乳,静置后,用0.22 μm有机系微孔滤膜滤过,取续滤液,按“2.2.3”项下色谱条件测定藤黄酸与雷帕霉素药物含量。按下列公式分别计算包封率和载药量。
包封率=药物实际质量/药物的投入量
载药量=药物实际质量/制剂的总质量
2.5 GR@Lip处方工艺优化
在文献调研及前期预实验的基础上,确定以制备温度(旋蒸及水化时的温度)、药脂比(脂总量代表磷脂与胆固醇总量)、磷脂-胆固醇比例、超声功率、超声时间、水化介质及水相pH值作为考察对象,包封率、载药量及粒径作为评价指标进行单因素考察,并根据单因素筛选结果,进行正交设计试验,筛选出最佳处方。
2.5.1 单因素考察
(1)制备温度考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,药脂比为1∶1∶15,磷脂-胆固醇比例为4∶1,超声功率195 W,超声时间5 min,超纯水作为水化介质,水相pH值为7.1的条件下,考察不同温度对包封率、载药量及粒径的影响。结果见表4,随着温度升高,包封率和载药量呈先上升后下降的趋势,综合考虑最佳制备温度为40 ℃,这可能是由于温度较低时无法引发磷脂相变,温度过高时影响磷脂稳定性造成的[10]。

表4 制备温度的考察(, n = 3)
(2)药脂比考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,磷脂-胆固醇比例为4∶1,超声功率195 W,超声时间5 min,超纯水作为水化介质,水相pH值为7.1的条件下,考察药脂比对GR@Lip包封率、载药量及粒径的影响。结果见表5,随着脂质用量的增加,藤黄酸和雷帕霉素的包封率逐渐增加,藤黄酸的载药量呈逐渐下降趋势,而雷帕霉素的载药量呈现先增加后下降的趋势,不同药脂比制得的脂质体粒径均小于200 nm,综合考虑最佳药脂比为1∶1∶20。这是由于增加脂质用量有利于增加包封率,但脂质用量较多时会降低载药量[11]。
(3)磷脂与胆固醇比例考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,药脂比为1∶1∶20,超声功率195 W,超声时间5 min,超纯水作为水化介质,水相pH值为7.1的条件下,考察药脂比对GR@Lip包封率、载药量及粒径的影响。结果见表6,随着磷脂比例的增加,藤黄酸和雷帕霉素的包封率略有下降,载药量先增加后下降,粒径逐渐增加,但均小于200 nm,综合考虑磷脂与胆固醇的最佳比例为4∶1。
表5 药脂比考察(, n = 3)
(4)超声功率考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,药脂比为1∶1∶20,磷脂与胆固醇比例4∶1,超声时间5 min,超纯水作为水化介质,水相pH值为7.1的条件下,考察超声功率对GR@Lip包封率、载药量及粒径的影响。结果见表7,随着功率的增加,藤黄酸和雷帕霉素的包封率和载药量逐渐增加,粒径逐渐减小。综合考虑,确定超声功率为195 W。
表6 磷脂与胆固醇比例考察(, n = 3)
表7 超声功率考察(, n = 3)
(5)超声时间考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,药脂比为1∶1∶20,磷脂与胆固醇比例4∶1,超声时间功率195 W,超纯水作为水化介质,水相pH值为7.1的条件下,考察超声时间对GR@Lip包封率、载药量及粒径的影响。结果见表8,随着超声时间的增加,藤黄酸和雷帕霉素的载药量和粒径逐渐减小,这可能是由于过长时间的超声破坏了GR@Lip。超声5 min时的包封率、载药量及粒径均较为理想。综合考虑,确定超声时间为5 min。
表8 超声时间考察(, n = 3)
(6)水化介质考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,药脂比为1∶1∶20,磷脂与胆固醇比例4∶1,超声时间功率195 W,超声时间5 min,水相pH值为7.1的条件下,考察水化介质对GR@Lip包封率、载药量及粒径的影响。结果见表9,选择超纯水作为水化介质时,包封率及载药量均明显优于PBS和生理盐水,且粒径较小。此外,生理盐水作为水化介质时,静置数小时便析出沉淀,表明其稳定性较差。综合考虑,确定超纯水作为水化介质。
(7)水相pH值考察:藤黄酸投药量为5 mg,雷帕霉素投药量为7.28 mg,温度为40 ℃,药脂比为1∶1∶20,磷脂与胆固醇比例4∶1,超声时间功率195 W,超声时间5 min,超纯水作为水化介质的条件下,考察水相pH值对GR@Lip包封率、载药量及粒径的影响。结果见表10,当水化介质呈弱酸性和近中性时,包封率及载药量均较高,呈弱碱性时略低,这可能是因为藤黄酸呈酸性,在碱性溶液中不稳定。在弱酸性条件下,共载脂质体的粒径略大。综合考虑,确定水相pH值为7.1。
表9 水化介质考察(, n = 3)

表10 水相pH值考察(, n = 3)
2.5.2 正交设计试验 综合比较单因素实验筛选结果,选择对包封率和载药量影响较为显著的3个因素,即制备温度(A)、药脂比(B)、磷脂-胆固醇比例(C)作为正交设计试验的主要影响因素,其他因素采用单因素考察的最佳结果,确定超声功率为195 W,超声时间为5 min,水化介质为超纯水,水相pH为7.1。在3个主要因素水平上以藤黄酸包封率(1)、雷帕霉素包封率(2)、藤黄酸载药量(3)、雷帕霉素载药量(4)及粒径(5)作为主要评价指标,采用L9(34)正交试验表进行实验,筛选出最优处方,正交因素设计水平见表11。
本研究因有多个评价指标,故采用综合评分法。通过SPSS AUA分析软件确定藤黄酸包封率(1)、雷帕霉素包封率(2)、藤黄酸载药量(3)、雷帕霉素载药量(4)及粒径(5)权重分别为20.10%、18.81%、18.22%、17.59%和25.27%,进行综合评分。正交设计试验结果见表11,方差结果见表12。极差为A>B>C,说明A因素的影响最大,即制备温度,其次是药脂比,磷脂与胆固醇的比例的影响较小,最佳制备工艺为A1B2C2,即GR@Lip的最优处方为制备温度40 ℃,药脂比1∶1∶20,磷脂与胆固醇比例4∶1。

表11 正交试验设计与结果

表12 方差结果分析
2.5.3 处方验证 根据正交设计试验结果,筛选出最优处方并平行制备3组,测定其包封率、载药量及粒径,对处方工艺进行验证。结果显示,GR@Lip中藤黄酸包封率为(97.27±2.76)%,雷帕霉素包封率为(96.58±3.82)%,藤黄酸载药量为(3.29±0.44)%,雷帕霉素载药量为(4.91±0.44)%,平均粒径为(157.19±1.74)nm,结果表明该方法重复性良好。
2.6 GR@Lip的表征及稳定性
2.6.1 表征 通过纳米粒径及ζ电位分析仪测定GR@Lip的粒径和ζ电位,采用TEM对GR@Lip进行形貌表征,考察GR@Lip在4 ℃条件下放置1个月的粒径和PDI变化情况。结果见图2和表13,GR@Lip的平均粒径为(157.19±1.74)nm,PDI为0.224±0.033,ζ电位为(−22.1±1.3)mV。TEM图像显示,共载药脂质体呈球形形态,在4 ℃条件下放置1个月粒径及PDI均无明显变化,表明脂质体的粒径具有良好的稳定性。
2.6.2 稳定性
(1)包封率及渗漏率的测定:将制备得到的GR@Lip放置于在4 ℃条件下,每3天通过HPLC测定包封率,并计算渗漏率(渗漏率=1-储存过程中测得的药物包封率/第0天测得的药物包封率)。结果如表14所示,随着储存时间延长,脂质体存在一定程度的泄露,但储存15 d包封率仍大于80%,符合脂质体质量控制标准。
(2)氧化产物的测定:磷脂氧化程度是脂质体稳定性的重要指标,本研究采用氧化产物评价磷脂的氧化程度。由于磷脂中含不饱和类脂,可被氧化为共轭二烯,形成过氧化脂质,进一步分解为丙二醛及溶血磷脂。丙二醛在酸性条件下与硫代巴比妥酸(thiobarbituric acid,TBA)反应生成一种红色染料(TBA-pigment),在532 nm处有最大吸收峰,吸光度()值大小可以反映磷脂的氧化程度。因此,将制备得到的GR@Lip放置于在4 ℃条件下,每3天通过丙二醛检测试剂盒测定532 nm处的值。结果如表15所示,GR@Lip在15 d内值无明显变化,表明脂质体具有良好的稳定性。
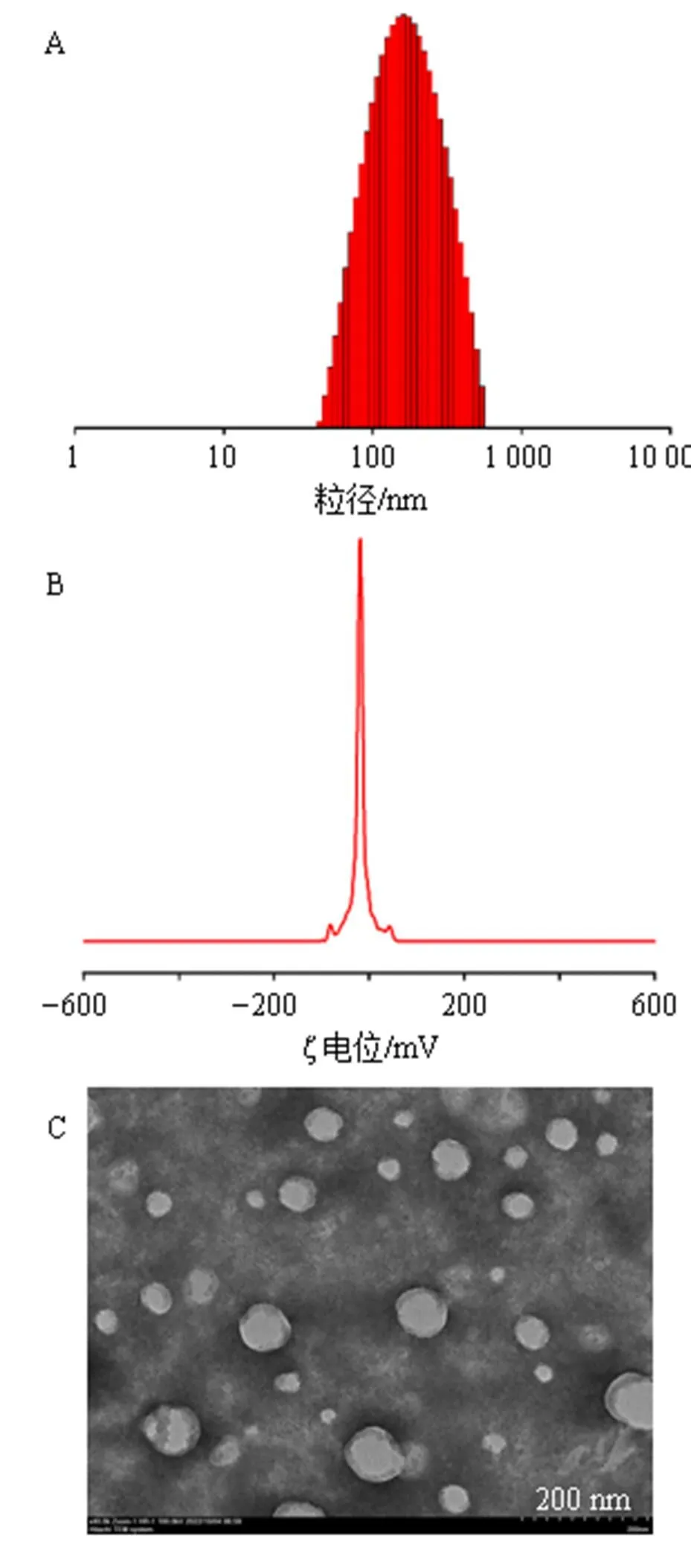
图2 GR@Lip的粒径分布(A)、ζ电位(B) 和TEM图(C)
表13 粒径稳定性结果(, n = 3)
表14 包封率及渗漏率测定结果(, n = 3)
表15 氧化产物测定结果(, n = 3)
2.7 体外抗肿瘤活性
2.7.1 细胞增殖毒性检测 通过CCK-8法测定GR@ Lip对3种不同癌细胞的增殖毒性,将处于对数生长期的Pan02、U14及4T1细胞以5 000个/孔接种在96孔板中,孵育24 h后弃去旧培养基,加入不同质量浓度的脂质体(GA@Lip、Rap@Lip、GR@ Lip)继续培养24 h,然后加入CCK-8增殖检测试剂,37 ℃孵育2 h后,通过酶标仪测定450 nm处的值,并计算IC50及CI。结果如表16所示,与GA@Lip组和Rap@Lip组相比,GR@Lip处理后3种癌细胞的IC50均显著降低,分别为(0.37+0.54)、(0.54+0.78)、(0.34+0.48)μg/mL。此外,GR@Lip对Pan02、U14及4T1细胞的CI值分别为0.65±0.01、0.96±0.02、0.86±0.01,均小于1,表明藤黄酸与雷帕霉素联用具有协同作用。
表16 IC50及CI值计算结果(, n = 3)
2.7.2 细胞凋亡检测 将处于对数生长期的Pan02、U14及4T1细胞以6×106个/孔接种在6孔板中,孵育24 h后弃去旧培养基,PBS洗涤。加入药物继续培养24 h,用PBS清洗后消化细胞,离心,收集细胞。加入500 μL的binding buffer(1×)重悬细胞,加入2.5 μL的Annexin-V-FITC和2.5 μL的PI染色液轻轻混匀染色,避光孵育15 min后置于冰上,1 h内用流式细胞仪检测。结果见图3和表17,在Pan02、U14及4T1细胞中,与对照组的凋亡率[(4.20±0.42)%、(4.03±0.46)%、(1.95±0.30)%]及单药组的凋亡率[GA@Lip:(22.24±0.22)%、(9.28±0.35)%、(15.45±1.79)%;Rap@Lip: (13.38±0.76)%、(11.95±0.42)%、(0.52±0.13)%]相比,GR@Lip的凋亡率分别为(32.56±1.44)%、(22.15±0.44)%和(25.62±1.58)%,均显著高于其他3组,表明藤黄酸与雷帕霉素联用具有促进肿瘤细胞凋亡的协同作用。

图3 GR@Lip对Pan02 (A)、U14 (B) 及4T1 (C) 细胞凋亡的影响

表17 细胞凋亡分析(, n = 3)
与对照组比较:***<0.001。
***< 0.001control group.
2.7.3 细胞迁移能力检测 通过细胞划痕实验研究对细胞迁移的影响,将处于对数生长期的Pan02、U14及4T1细胞,以8×106个/孔接种在12孔板中,孵育24 h后弃去旧培养基,PBS清洗细胞后,采用200 μL移液枪枪头在单层细胞上划一字划痕,PBS清洗,在倒置荧光显微镜下观察并拍照,记录为0 h。随后加入药物继续培养24 h,PBS清洗1次后,在倒置荧光显微镜下拍照观察细胞的迁移情况。使用Image J软件分析划痕面积,并计算迁移率[迁移率=(0 h划痕面积-24 h划痕面积)/0 h划痕面积]。结果见图4和表18,在无药物干预的情况下,左右两侧细胞逐渐向中间划痕区域靠拢,Pan02、U14及4T1细胞的迁移率达到(51.86±2.35)%、(29.37±8.57)%和(46.96±2.94)%,而给药组向划痕区域迁移情况均受到一定程度抑制,尤其给予GR@Lip后,各组细胞迁移率分别降至(3.21±1.01)%、(5.98±2.69)%和(12.39±6.43)%,且显著低于单药组,表明藤黄酸与雷帕霉素联用具有协同抑制肿瘤细胞迁移的作用。

图4 GR@Lip对Pan02 (A)、U14 (B) 及4T1 (C) 细胞迁移的影响
表18 细胞迁移分析(, n = 3)
与对照组比较:**<0.01***<0.001。
**< 0.01***< 0.001control group.
2.7.4 细胞侵袭能力检测 通过Transwell实验检测对细胞侵袭的影响,将Matrigel基质胶与无血清培养基,分别按1∶8、1∶12、1∶14的比例进行稀释,以100 μL/孔加到Transwell上室中,置于培养箱中恒温孵育5 h至其完全凝固。吸去多余液体,将处于对数生长期的Pan02、U14及4T1细胞分别以2×104、5×104、6×104个/孔接种于Transwell上室,并加入100 μL含药无血清培养基,下室加入750 μL的完全培养基,继续培养24 h,弃去Transwell上室的培养基,4%多聚甲醛固定30 min,PBS洗涤,0.5%结晶紫染色5 min。染色结束后,PBS漂洗3次,自然晾干,于显微镜下选取随机视野,观察并拍照。结果见图5,与对照组相比,给药组穿过Transwell小室聚碳酸酯膜的细胞数量显著减少,其中GR@Lip组细胞数量最少,表明藤黄酸与雷帕霉素联用可以发挥抑制肿瘤细胞侵袭的协同作用。

图5 GR@Lip对Pan02 (A)、U14 (B) 及4T1 (C) 细胞侵袭的影响
2.7.5 自噬小体检测 通过TEM观察GR@Lip对诱导自噬的影响。将处于对数生长期的Pan02、U14及4T1细胞以1.5×106个/孔接种在60 mm培养皿中,孵育24 h后弃去旧培养基,PBS清洗,加入药物继续培养24 h。除去旧培养基,PBS洗涤1次,用胰酶消化,收集细胞悬液至15 mL离心管中,1 500 r/min离心(离心半径为16.8 cm)10 min,弃去上清。
加入0.5%戊二醛固定液重悬,4 ℃静置10 min, 12 000 r/min离心(离心半径为8.6 cm)15 min,弃去上清。加入3%戊二醛固定液固定,1%四氧化锇进行再固定,丙酮逐级脱水,Epon812包埋,半薄切片用甲苯胺蓝染色作光学定位,用钻石刀作超薄切片,醋酸铀和枸橼酸铅染色后通过透射电镜观察并拍照。结果见图6,与对照组相比,药物干预后各组均可观察到自噬小体的产生(红色箭头),其中GR@Lip组自噬小体数量最多,表明藤黄酸与雷帕霉素联用可以协同增强自噬。

图6 GR@Lip对Pan02 (A)、U14 (B) 及4T1 (C) 自噬小体形成的影响
2.7.6 自噬相关蛋白LC3B和Beclin1的表达 采用免疫荧光技术检测自噬相关蛋白LC3B和Beclin1的表达情况。将处于对数生长期的Pan02、U14及4T1细胞,以1×105个/孔接种在铺好细胞爬片的12孔板中,孵育24 h后弃去旧培养基并用PBS清洗,加入药物继续培养24 h后,用4%多聚甲醛固定。结果见图7,GA@Lip和Rap@Lip处理后,在3种癌细胞中均可观察到红色荧光(LC3B)或绿色荧光(Beclin1)有所增强,表明藤黄酸与雷帕霉素均可激活自噬。同时,GR@Lip组中红色荧光和绿色荧光均显著增强,表明藤黄酸与雷帕霉素联合可以协同诱导自噬。
2.8 体内药动学研究
2.8.1 给药方案与样品处理 Balb/c小鼠购进后适应性喂养1周,随机分为空白组、游离藤黄酸组4 mg/kg、游离雷帕霉素组6 mg/kg、GR@Lip组(4+6)mg/kg,尾静脉给药1次后,分别于0.25、0.50、2.00、4.00、8.00、12.00、24.00 h通过小鼠眼球取血,3 500 r/min离心15 min(离心半径为8.6 cm),取上层血浆于−80 ℃保存。
2.8.2 内标储备液与对照品储备液的配制 精密称取内标物质和厚朴酚、子囊霉素和对照品藤黄酸、雷帕霉素各5 mg,于10 mL量瓶中,加乙腈溶解并稀释至刻度,作为内标物质储备液和对照品储备液。
(1)内标溶液配制:精密量取和厚朴酚内标储备液20 μL、子囊霉素内标储备液20 μL到960 μL乙腈中,混匀,再移取50 μL到24.95 mL乙腈中,配制成20 ng/mL内标溶液,4 ℃贮存。
(2)标准曲线建立:精密量取藤黄酸和雷帕霉素对照品储备液,分别用乙腈稀释为1 000、400、100、40、10、4、1 ng/mL,作为对照品溶液。按色谱条件进样测定,分别以雷帕霉素、藤黄酸的质量浓度为横坐标(),以雷帕霉素、藤黄酸峰面积与内标物质峰面积比值为纵坐标()绘制标准曲线,得回归方程分别为雷帕霉素=8 798.467 5+ 5 691.288 7,2=0.992 0;藤黄酸=23 967.095 9-11 876.545 8,2=0.994 7。
2.8.3 色谱条件 色谱柱为Waters UPLC BEH C18柱(100 mm×2.1 mm,1.7 μm);流动相为乙腈-0.01%氨水溶液,梯度洗脱:0~1.00 min,40%~90%乙腈;1.00~2.80 min,90%乙腈;柱温35 ℃;进样量1 µL;体积流量0.5 mL/min。
2.8.4 质谱条件 电离方式:电喷雾离子源,离子源温度500 ℃,气帘气(CUR)137.895 kPa(20 psi),喷雾电压(IS)−4 500 V,雾化气(GS1)413.685 kPa(60 psi),加热气(GS2)413.685 kPa(60 psi),去簇电压(DP)−100 V,离子束聚焦电压(EP)−10 V,碰撞池出口电压(CXP)−10 V,总扫描时间0.315 s,监测模式:负离子多反应监测(MRM)。定量分析的离子反应对为藤黄酸/627.2→583.2,碰撞能−22 V;雷帕霉素/912.6→321.2,碰撞能−49 V。
2.8.5 液质联用分析 精密量取30 μL待测血浆样品,加入120 μL含和厚朴酚和子囊霉素20 ng/mL的内标溶液,涡旋混匀,13 000 r/min离心15 min(离心半径为8.6 cm),取上清液至进样小瓶,采用上述色谱、质谱条件进行含量检测。
2.8.6 药动学结果 根据上述检测结果,绘制血药浓度-时间曲线见图8,采用DAS 2.0软件对所得数据进行计算分析,得出相关药动学参数见表19。与游离药物组相比,GR@Lip组中藤黄酸和雷帕霉素的AUC0~t分别是游离藤黄酸和游离雷帕霉素的1.51和1.29倍,AUC0~∞分别是游离藤黄酸和游离雷帕霉素的1.64和1.34倍。
藤黄酸和雷帕霉素的达峰浓度(max)分别由(27.29±4.61)μg/L和(148.19±5.07)μg/L提高至(36.43±1.53)μg/L和(179.69±25.78)μg/L。更重要的是,藤黄酸和雷帕霉素的半衰期1/2分别由3.67 h和6.82 h延长至8.48 h和8.12 h,且平均滞留时间(MRT0~∞)较游离组也有所延长。以上结果表明GR@Lip较游离药物可在体内滞留更长时间,具有更高的生物利用率。
2.9 体内生物分布
采用荧光染料ICG代替药物进行脂质体的体内生物分布研究,Balb/c小鼠购进后适应性喂养1周,将处于对数生长期的4T1细胞以3×105个/100 μL接种于小鼠第2乳腺垫皮下,建立小鼠乳腺癌原位模型。待肿瘤生长至200 mm3时,随机分为2组,静脉给予1 μg/mL的ICG Lip与游离ICG,分别于1、2、4、6、8、10、24、48 h置于活体成像仪拍摄。待小鼠体内荧光染料完全清除后(大于72 h),再次通过尾静脉给予相同剂量的ICG@Lip与游离ICG,于8 h时处死小鼠,解剖出心、肝、脾、肺、肾及肿瘤组织,采用4%多聚甲醛固定,进行离体成像,分析各组织中ICG的荧光强度。结果见图9,随着时间的推移,游离ICG组中肿瘤部位的ICG荧光强度逐渐减弱且下降速度较快,24 h时荧光几乎完全消失。而ICG@Lip组的ICG荧光强度下降速度相对缓慢,且从4 h起显著高于游离ICG组,甚至在48 h时仍旧保留部分荧光。

图7 GR@Lip对Pan02 (A)、U14 (B) 及4T1 (C) 细胞中自噬相关蛋白LC3B和Beclin1表达的影响

图8 血药浓度-时间曲线(, n = 3)
心、肝、脾、肺、肾和肿瘤组织的离体成像结果显示,ICG@Lip组在肿瘤部位的荧光明显强于游离ICG组。上述结果显示,与游离ICG相比,ICG@ Lip可更长时间富集于肿瘤部位,表明脂质体可通过被动靶向改善药物在体内的分布,增强肿瘤靶向性,增加蓄积量,这可能依赖于纳米颗粒的高通透性和滞留(enhanced permeability and retention effect,EPR)效应。
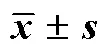
表19 主要药动学参数(, n = 3)
与游离雷帕霉素比较:*<0.05**<0.01***<0.001。
*< 0.05**<0.01***< 0.001free rapamycin.
3 讨论
脂质体制备方法通常包括溶剂注入法、薄膜水化法和逆向蒸发法等。本研究首先考察了3种不同制备方法对GR@Lip包封率、载药量及粒径的影响,确定薄膜水化法为GR@Lip的最佳制备方法,并进一步通过单因素实验和正交设计试验进行筛选,得到了最优的处方工艺,实现了2药的高效包载。然而,薄膜水化法制备所得脂质体通常粒径较大且不均一[12],影响脂质体的稳定性,而脂质体的稳定性决定了其在体内的循环时间与到达作用靶点的药物量,从而对药物的治疗效果产生重要影响[13]。因此,在水化后采用超声破碎方式使脂质体的平均粒径控制在200 nm以内,能在30 d内维持粒径和PDI基本不变化,改善了该制备方法的不足,并通过考察渗漏率和磷脂氧化程度证明了其具有良好的稳定性。此外,在藤黄酸检测方法[14]的基础上进行了改进,建立了一种准确度高、可靠性强的HPLC分析方法,可用于同时检测藤黄酸和雷帕霉素的含量。

A-不同时间点下荷瘤小鼠的活体荧光图;B-8 h后离体脏器及肿瘤荧光图;C-肿瘤部位荧光强度随时间变化曲线(, n = 3, *P<0.05 ***P<0.001)。
实体瘤具有高度异质性,单一药物的疗效有限,加大药物剂量易带来不良反应以及多药耐药性等问题[15],联合疗法可以通过作用不同通路以及不同靶点,减少耐药的发生,并可通过药物的协同作用进一步提高药物疗效。基于肿瘤病理复杂性,抗肿瘤药物联合使用已成为临床上的主要策略[16]。本研究将天然化合物藤黄酸与mTOR抑制剂雷帕霉素联用,实现了协同抗胰腺癌、宫颈癌及乳腺癌的作用,与空白对照及单药组相比,GR@Lip在诱导凋亡、抑制迁移和侵袭方面均展现出显著优势。此外,研究表明自噬在控制癌症的发生发展,影响肿瘤细胞对抗癌治疗反应中发挥着重要的作用,诱导肿瘤细胞自噬已被证明是有效的干预措施[17]。雷帕霉素作为一种经典的自噬激活剂,可负性调控PI3K/Akt/ mTOR通路,激活自噬介导的死亡。研究表明,藤黄酸也能够通过上调自噬蛋白Beclin1、Atg和LC3的表达,诱导自噬小体的形成[18]。
此外,大量证据表明Akt/mTOR信号通路的抑制可激活自噬关键调节因子Beclin1[19],这表明藤黄酸可能上调Beclin1和LC3B的表达协同雷帕霉素抑制Akt/mTOR信号通路,从而增强肿瘤细胞自噬。本研究结果也证实藤黄酸和雷帕霉素可协同促进肿瘤细胞产生自噬。有研究报道,雷帕霉素可诱导细胞产生自噬性凋亡[20],雷帕霉素单独使用时,肿瘤细胞仅发生少量凋亡,联合应用时凋亡率显著增加。这提示藤黄酸联合雷帕霉素对肿瘤细胞的凋亡作用可能是通过协同增强自噬介导的,但具体机制需要后续的实验进一步研究。
综上,本研究采用薄膜水化法制备藤黄酸和雷帕霉素共载脂质体GR@Lip,并进行体外抗肿瘤作用、体内药代动力学及生物分布研究。结果表明GR@Lip在胰腺癌、宫颈癌及乳腺癌细胞模型上可通过抑制肿瘤细胞增殖、诱导凋亡、抑制迁移和侵袭以及激活自噬等多种途径发挥协同抗肿瘤作用,与游离药物相比在体内和肿瘤部位可滞留更长时间,显示出用于治疗恶性肿瘤的潜力,为后续体内抗肿瘤研究奠定了基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 徐振娜, 赵逸卿, 陈思宇, 等. 中西药联合用药的优势及风险分析 [J]. 中草药, 2023, 54(2): 408-415.
[2] Chen Z J, Hong G H, Liu Z Y,. Synergistic antitumor efficacy of doxorubicin and gambogic acid-encapsulated albumin nanocomposites [J]., 2020, 196: 111286.
[3] Hatami E, Nagesh P K B, Jaggi M,. Gambogic acid potentiates gemcitabine induced anticancer activity in non-small cell lung cancer [J]., 2020, 888: 173486.
[4] Liu Y L, Chen Y C, Lin L F,. Gambogic acid as a candidate for cancer therapy: A review [J]., 2020, 15: 10385-10399.
[5] Chen X M, Chen D R, Liu H M,. Local delivery of gambogic acid to improve anti-tumor immunity against oral squamous cell carcinoma [J]., 2022, 351: 381-393.
[6] 赖梦琴, 张鹏, 杨明, 等. 吉西他滨单磷酸盐/紫杉醇联用靶向纳米粒的制备及其大鼠体内药动学研究 [J]. 中草药, 2021, 52(3): 669-676.
[7] Kiaie S H, Mojarad-Jabali S, Khaleseh F,. Axial pharmaceutical properties of liposome in cancer therapy: Recent advances and perspectives [J]., 2020, 581: 119269.
[8] 彭剑青, 邹颖, 徐金转, 等. 共载光敏剂/UCNPs/双氢青蒿素脂质体的制备及其体外抗肿瘤活性评价 [J]. 中草药, 2020, 51(16): 4151-4159.
[9] 田文源, 陈飞虹. IDO1抑制剂联合替莫唑胺对人脑胶质瘤的协同抗肿瘤作用研究 [J]. 药学学报, 2022, 57(3): 707-715.
[10] 刘万路. 牛血清白蛋白/壳聚糖双层包覆染料木素脂质体的制备、表征和口服药动学研究 [J]. 中草药, 2023, 54(24): 8018-8030.
[11] 张体鹏, 决利利. 白屈菜红碱2种脂质体的制备和口服生物利用度比较 [J]. 中草药, 2023, 54(17): 5568-5579.
[12] Maja L, Željko K, Mateja P. Sustainable technologies for liposome preparation [J]., 2020, 165: 104984.
[13] 金彤彤, 陆雯沁, 张傅瑜淇, 等. 脂质体稳定性影响因素的研究 [J]. 光谱学与光谱分析, 2023, 43(S1): 287-288.
[14] 王蕊, 杨大宇, 许贵军, 等. HPLC法测定含藤黄不同外用制剂中的藤黄酸含量 [J]. 化学工程师, 2020, 34(4): 24-27.
[15] 江文心, 张华清, 丁杨, 等. 抗肿瘤多药联用型纳米递送系统的研究进展 [J]. 药学学报, 2022, 57(1): 1-12.
[16] Yu D S, Wang Y Z, Chen J F,. Co-delivery of NIR-II semiconducting polymer and pH-sensitive doxorubicin-conjugated prodrug for photothermal/chemotherapy [J]., 2022, 137: 238-251.
[17] Ichimiya T, Yamakawa T, Hirano T,. Autophagy and autophagy-related diseases: A review [J]., 2020, 21(23): 8974.
[18] Kashyap D, Mondal R, Tuli H S,. Molecular targets of gambogic acid in cancer: Recent trends and advancements [J]., 2016, 37(10): 12915-12925.
[19] Wang H C, Zhao Z, Lei S Z,. Gambogic acid induces autophagy and combines synergistically with chloroquine to suppress pancreatic cancer by increasing the accumulation of reactive oxygen species [J]., 2019, 19: 7.
[20] 王亚奇, 高晓娟, 岳保红. Nucleostemin下调联合雷帕霉素对HL-60细胞自噬与凋亡影响的初步研究 [J]. 中国实验血液学杂志, 2023, 31(6): 1629-1634.
Preparation of gambogic acid and rapamycin co-loaded liposomes and study on the synergistic antitumor effect
DING Huanhuan1, 2, JIANG Xianmei2, 3, LIU Hongmei2, 3, QI Yao2, 3, TU Kerong2, 3, CHEN Lin1, CAI Lulu2, 3, ZHOU Xianli1
1. School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China 2. Department of Pharmacy, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s hospital, Chengdu 610072, China 3. Personalized Drug Therapy Key Laboratory of Sichuan Province, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China
To prepare liposomes co-loaded with gambogic acid and rapamycin (GR@Lip), optimize its prescription, perform anti-tumor mechanism research, pharmacokinetics and biodistribution.The entrapment efficiency, drug loading, particle size were used as evaluation indicators. Single factor investigation method and orthogonal design experiments were used to optimize the optimum formulation of GR@Lip, and its characterization and stability were studied. The effects of GR@Lip on the proliferation and apoptosis of tumor cells were investigated by CCK-8 and flow cytometry. The effects of GR@Lip on the migration and invasion of tumour cells were investigated by cell scratching, Transwell. The effects of GR@Lip on the autophagy of tumour cells were investigated by transmission electron microscope (TEM) and immunofluorescence. The effects of GR@Lip on the pharmacokinetics and biodistributionwere investigated by liquid chromotography with mass spectrometry (LC-MS) and living body imager.The optimal conditions were determined as follows: temperature is 40 ℃, ratio of drug to lipid is 1:1:20, the phospholipid-cholesterol ratio is 4:1, ultrasonic power is 195 W, ultrasonic time is 5 min, hydration medium is ultrapure water and aqueous pH value is 7.1. The entrapment efficiency and drug loading of gambogic acid and rapamycin were (97.27 ± 2.76)%, (96.58 ± 3.82)%, (3.29 ± 0.44)% and (4.91 ± 0.44)%, respectively. GR@Lip showed a spherical under TEM with an average particle size of (157.19 ± 1.74) nm, ζ potential was (−22.1 ± 1.3) mV by dynamic light scattering (DLS), and had good stability.anti-tumor activity experiment showed that gambogic acid combination rapamycin can inhibit the proliferation, migration and invasion of tumor cells, and also significantly promote apoptosis and autophagy. In addition, pharmacokinetics and biodistribution resultsshowed that GR@Lip can remain for long time and had good tumor targeting.This study successfully prepare GR@Lip, revealing that it has synergistic anti-tumor activity through multiple pathways, and significantly improving the pharmacokinetic behavior of gambogic acid and rapamycin, providing a basis for furtherresearch and future clinical applications.
gambogic acid; rapamycin; co-loaded liposomes; anti-tumor; drug combination;pharmacokinetics;biodistribution;proliferation; apoptosis; migration; invasion; autophagy
R283.6
A
0253 - 2670(2024)09 - 2896 - 16
10.7501/j.issn.0253-2670.2024.09.005
2023-10-10
国家自然科学基金项目(U2230123);国家自然科学基金项目(82304969);国家自然科学基金项目(82304789);国家自然科学基金项目(81972901);四川省科技项目(2022ZYD0080);四川省科技项目(2023YFS0110);四川省科技项目(2023YFS0131);四川省科技项目(2023YFS0125);四川省科技项目(2023NSFSC0033);中国博士后科学基金(2022M710623);中国博士后科学基金(2022M720670)
丁欢欢,硕士研究生,研究方向为药剂学。E-mail: 2467091987@qq.com
通信作者:周先礼,教授,研究方向为天然药物化学。E-mail: zhouxl@swjtu.edu.cn
蔡璐璐,教授,研究方向为肿瘤靶向药物制剂研究。E-mail: cailulu@med.uestc.edu.cn
[责任编辑 郑礼胜]
猜你喜欢
——雷帕霉素
食品与健康(2022年8期)2022-10-22
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
骨科临床与研究杂志(2020年6期)2020-03-02
临床医药文献杂志(电子版)(2020年23期)2020-02-28
海峡科技与产业(2016年3期)2016-05-17
吉林大学学报(医学版)(2015年3期)2015-12-17
小说月刊(2015年6期)2015-12-16
安徽中医药大学学报(2014年2期)2014-06-19
长江大学学报(自科版)(2014年23期)2014-03-27
