
桥粒胶蛋白2及其在肿瘤疾病中的研究进展
2024-05-10 01:47魏盈盈徐敏娟谢水祥王烈峰
赣南医学院学报 2024年3期
魏盈盈,徐敏娟,谢水祥,王烈峰
(1.赣南医科大学基础医学院;2.赣州市人民医院妇产科,江西 赣州 341000)
上皮细胞存在几种类型的细胞连接,可分为黏附连接、紧密连接、桥粒和间隙连接,它们将组织中的细胞进行连接,并在组织屏障功能、细胞增殖和迁移过程中调节组织的平衡。细胞连接的缺陷引起了广泛的组织异常,破坏了内稳态,在遗传异常和癌症中很常见[1]。研究发现,细胞黏附成分对肿瘤细胞的侵袭和转移非常重要,这不仅与细胞黏附变化有关,还参与了关键的分子信号通路[2]。桥粒(Desmosome,DSM)是一种细胞间锚定连接结构,对于维持经历机械应力组织(如皮肤、心肌和胃肠道黏膜)的完整性至关重要。桥粒是一种黏连方式,将细胞骨架中的中间丝锚定在细胞表面。桥粒胶蛋白(Desmocollins,DSCs)属于钙黏蛋白超基因家族,在细胞间黏附中发挥重要作用[3]。与经典钙黏蛋白相比,桥粒钙黏蛋白家族可以分为2种类型:桥粒芯蛋白(Desmogleins,DSG 1~4)和桥粒胶蛋白(Desmocollins,DSC 1~3),它们以细胞类型和分化依赖的方式表达[4]。DSG3和DSC3是在基底表皮中表达的主要亚型,基底表皮是寻常型天疱疮形成水疱的部位。而DSG1和DSC1主要在表皮浅表皮中表达,在基底层中几乎没有表达[5]。DSC2介导细胞间黏附并参与肿瘤进展[6]。DSC2对上皮细胞的统一性至关重要,并在上皮细胞的形态发生、分化、伤口愈合、细胞凋亡、迁移和增殖等过程中充当重要的调节器[7]。本文综述了DSC2的结构和功能,探讨了DSC2的分子机制及其表达异常与肿瘤的关系。
1 DSC2功能与作用机制
1.1 DSC2的分子结构桥粒是连接相邻细胞角蛋白中间丝的细胞间连接结构[8]。桥粒可锚定角蛋白中间丝并在细胞之间提供强大的黏附力,因此是维持组织结构和完整性的关键[9]。桥粒由许多蛋白质组成,包括桥粒钙黏蛋白、犰狳蛋白和血小板亲和蛋白,它们负责介导细胞间黏附[10]。桥粒胶蛋白(DSCs)细胞质尾部由细胞内锚定(Intracellular anchor,IA)和细胞内钙黏蛋白样序列(Intracellular cadherin-like sequence,ICS)组成[11]。其通过选择性剪接产生一个较长的“a”形式和一个较短的“b”形式[12]。已证明3个DSC基因(DSC1~3)和4个DSG基因(DSG1~4)具有类似的结构,DSCs和DSGs之间大约有30%的序列相同[13]。与所有的桥粒钙黏蛋白一样,人类DSC2基因位于18号染色体的长臂上,由32 kb以上的DNA组成,分为17个外显子,大小在46~258 bp之间。其中,第16个外显子可以选择性剪接,产生2种异构体,一个较长的“a”异构体DSC2a和一个较短的“b”异构体DSC2b。DSC2蛋白含有901个氨基酸,分子量为99 962 Da。DSC2是一种跨膜蛋白,包含细胞外结构域和细胞内结构域[7]。DSC2蛋白结构如图1所示。DSC2胞外结构域含有4个钙黏蛋白重复序列(Extracellular cadherin,EC)。每一个都有2个Ca2+结合位点,第5个结构域称为细胞外锚定(Extracellular anchor,EA)结构域[14]。在细胞质中,DSC2a的C端区域包含一个ICS,与斑珠蛋白(Plakoglobin,PG)或γ-catenin连接形成复合物锚,将中间丝与细胞膜结合,形成一个细胞外支架,从而抵抗机械应力。而DSC2b异构体不能在无ICS的情况下连接PG。因此,DSC2a与PG或γ-catenin的连接受损不能由DSC2b来补偿[7]。

图1 DSC2蛋白结构域
1.2 DSC2参与桥粒黏附脊椎动物中存在4种类型的细胞间连接:桥粒、黏附连接、紧密连接和间隙连接[15]。紧密连接选择性调节水、离子和溶质的细胞旁运动,而黏附连接介导细胞间黏附。黏附连接的存在是桥粒形成的先决条件,它们在细胞之间形成“点焊”,并具有增强细胞间黏附的功能[16]。桥粒不仅是静态同时也是高度动态的结构,它们是上皮细胞间黏附和支持上皮细胞动态重新排列的基础,能够在组织的形态发生和重塑过程中快速组装和拆卸。研究表明,桥粒膜和斑块的个别成分在初始阶段以可溶性成分的形式合成,然后通过空间和生化分隔转移,最终变得不溶并进入细胞膜形成桥粒[14]。桥粒黏附由桥粒芯蛋白(DSGs)和桥粒胶蛋白(DSCs)介导[16]。YULIS M等[17]提出,将桥粒中的DSC2进行荧光标记,并通过成像技术发现在整个细胞周期中,荧光标记点在细胞-细胞接触位点保持不动。然而,光漂白后荧光恢复实验证明,DSC2能够在必要时迅速做出反应并且能够重塑黏附结构[18]。在体外,通过延时荧光成像技术观察到桥粒钙黏蛋白DSG2和DSC2定位于不同的囊泡群,并在活细胞中独立移动到质膜[19]。
桥粒形成在2 h内分2个阶段发生,第1个阶段发生在转移到培养基的30 min内,在60 nm囊泡中,主要含有DSC2和较低浓度的DSG2及E-钙黏蛋白(E-cadherin,E-cad)分布到整个细胞表面,这些物质随后大部分被内吞。第2个阶段涉及DSG,E-cad、血红蛋白和β-catenin在更复杂的大小约200 nm的囊泡中转运,指向发育中的基底外侧表面上可能的成核位点,随后加入斑块蛋白,如桥粒蛋白Ⅰ/Ⅱ。第2个阶段的囊泡可以通过19 ℃预先标记的多囊体的逆行运输进入内吞标志物内[20]。桥粒钙黏蛋白DSCs和DSGs与由黏附连接的E-cad、P-钙黏蛋白(P-cadherin,P-cad)和N-钙黏蛋白(N-cadherin,N-cad)组成的经典钙黏蛋白家族密切相关[21]。DSCs和DSGs在细胞外结构域中具有钙结合口袋,使桥粒的从头组装为钙依赖性过程[16]。DSC2的缺失与细胞间连接组装过程中DSG2介导的桥粒张力变化有关,在钙耗竭和补充后,DSC2缺失导致钙转换后组装过程中DSG2靶向细胞间连接出现延迟,这表明DSC2有助于钙依赖性桥粒组装[22]。
DSC2在连接形成过程中控制桥粒介导的机械张力。DSC2缺失与桥粒斑蛋白Ⅰ/Ⅱ(DesmoplakinⅠ/Ⅱ,DPⅠ/Ⅱ)磷酸化的变化有关。DPⅠ/Ⅱ是一种桥粒支架蛋白(200~250 kDa),通过中间丝-细胞骨架与桥粒钙黏蛋白连接,在桥粒上产生张力[23]。角蛋白中间丝通过DP连接到DSC和桥粒芯蛋白DSG上[11]。DSC2通过调节DPⅠ/Ⅱ和细胞角蛋白结合,控制桥粒中的张力。DSC2缺乏导致细胞膜上DPⅠ/Ⅱ和角蛋白-中间丝的共定位增强,DPⅠ/Ⅱ和中间丝-细胞骨架之间的关联使DSC2缺乏在桥粒上产生更高的拉力[22]。
1.3 DSC2分子作用机制研究表明,DSC2的上调抑制了肝癌细胞中p-ERK1/2和c-MYC的表达,相应地,DSC2的下调促进了其表达,提示DSC2可能通过ERK/c-MYC信号通路调节肝癌细胞增殖、凋亡、迁移和侵袭[24]。影响肿瘤发生和发展的DSC2表达异常主要与β-catenin/γ-catenin信号通路有关。研究发现,DSCs通过激活β-catenin信号通路,促进肿瘤细胞的增殖和侵袭,从而促进恶性肿瘤的疾病进展[24]。DSC2通过E-cadherin依赖的方式抑制β-catenin信号传导来抑制细胞侵袭[25]。DSC2可通过对γ-catenin的影响间接调节β-catenin信号传导,γ-catenin是与DSCs相互作用的连环蛋白家族成员,当γ-catenin过量时,可能与E-cadherin“竞争”并取代β-catenin[26]。
KOLEGRAFF K等[26]研究发现,DSC2的缺失通过3种方式激活Akt/β-catenin信号通路促进细胞转化和增殖,如图2所示。⑴DSC2的下调导致结肠上皮细胞中表皮生长因子受体(Epidermal growth factor receptor,EGFR)激活和激磷脂酰肌醇3激酶(Phosphatidylinositol-3-kinase,PI3K)/Akt依赖性β-catenin/TCF转录激活。该途径的激活导致体内外细胞增殖和肿瘤生长增强。⑵AKT通过可磷酸化激活残基Ser-473和Thr-308的PH结构域被招募到质膜上富含磷脂酰肌醇-(3,4,5)-三磷酸(Phosphatidylinositol-3,4,5-triphosphate,PIP3)的区域。活化的Akt可通过在Ser-9处磷酸化含糖原合酶激酶3β(Glycogen synthase kinase 3β,GSK-3β)或 直 接 在Ser-552处磷酸化β-catenin来增强其信号。⑶在没有Wnt信号的情况下,β-catenin被复合物结合并靶向降解。在Wnt配体存在的情况下,Wnt与Fz/LRP受体结合,导致GSK-3的复合物与β-catenin解离,促进β-catenin积累[26]。DSC2的下调诱导EGFR激活,从而刺激PI3K活性并提高PIP3水平。Akt通过与PIP的相互作用被募集到膜中并被磷酸化激活。活化的Akt磷酸化并直接抑制GSK-3β和磷酸化β-catenin,增加β-catenin的核定位和转录活性。DSC2的缺失可能通过“孤立”或过量的DSG2导致Akt/β-catenin信号的激活[27]。
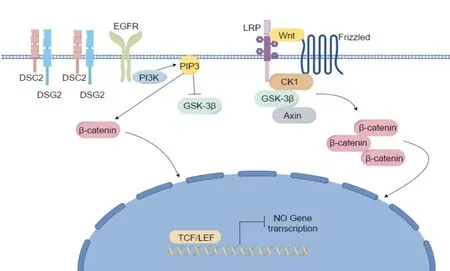
图2 AKT/β-catenin信号通路
2 DSC2与肿瘤的关系
超过90%的癌症起源于上皮细胞,因此上皮细胞之间的连接蛋白复合物如桥粒可能与癌症的进展有关。有研究[28]强调了桥粒在诱导癌症进展方面的潜在作用,特别是它们在癌症进展中的分解作用。桥粒是一种复杂的连接中间丝的细胞间连接结构,在暴露于机械应力的组织中提供强大的细胞间黏附力[29]。近年来,桥粒蛋白已成为癌症研究的一个关注点。通过对蛋白类型和原发性肿瘤定位发现,桥粒蛋白在肿瘤中表达水平的上调/下调对肿瘤的发生发展会产生增强/抑制作用[4]。
2.1 DSC2与结直肠癌桥粒复合物的失调与结直肠癌典型的组织结构紊乱有关[30]。肠道中的桥粒由黏附分子DSG2和DSC2组成,可通过增强细胞间 黏 附 力 而 促 进 黏 膜 稳 态[22,31]。肠 上 皮 细 胞(Intestinal epitheliel cells,IEC)中DSC2的缺失导致桥粒变小和桥粒内膜间隙增加,并导致体内肠上皮屏障缺陷,增加了体内肠道通透性,肯定了DSC2在调节肠上皮屏障功能中的作用[22]。GROSS A等[32]研究发现,与对照组小鼠相比,DSC2敲低小鼠的IEC屏障功能缺乏显著。IEC中DSC2的急性缺失导致在硫酸葡聚糖钠诱导的损伤中上皮恢复受损,表明DSC2在肠黏膜修复中具有保护作用[33]。DSC2通过调节整合素β1和β4蛋白以及Rap1活性来控制从IEC到细胞外间质的力的转导,而Rap1可以有效调节细胞迁移和伤口愈合能力[33]。DSC2在结直肠癌中的抑制可归因于同源框转录因子CDX1和CDX2(肠分化的常见调节因子)缺失。在结直肠癌中,DSC2缺失通过调节Akt/β-catenin信号通路促进肿瘤细胞增殖[26]。
2.2 DSC2与乳腺癌研究发现,多种类型的肿瘤细胞增殖和侵袭行为与DSC2异常表达相关,且DSC2水平高表达提高了细胞之间的聚集能力[34]。原发性乳腺癌组织中较高水平的DSC2显著影响HER2阳性和三阴性乳腺癌患者的疾病进展,LANDEMAINE T等[35]将DSC2鉴定为乳腺癌肺转移的潜在预测标志物。从功能上讲,肿瘤细胞中DSC2表达水平直接影响其聚集能力,进而影响其化学敏感性[36]。敲低桥粒蛋白会破坏乳腺癌细胞的聚集并降低失巢凋亡抗性[37]。DSG2和DSC2通过促进细胞聚集和提高肿瘤细胞传播过程中的存活率,从而提高乳腺癌细胞的转移能力[38-39]。DSC2在乳腺癌细胞中的上调导致细胞聚集能力增强,而DSC2沉默后的肿瘤细胞聚集体显示出更松散的结构,当受到机械应力时迅速解离[36]。
2.3 DSC2与食管癌在食管鳞状细胞癌(Esophageal squamous cell carcinoma,ESCC)中DSC2的表达从食管增生到不典型增生和原位癌逐渐降低[40]。FANG W K等[25]也发现DSC2在ESCC中表达降低,并且与肿瘤转移增强和预后不良有关。DSC2的过表达可以抑制ESCC的细胞增殖和转移。ESCC中DSC2下调导致游离γ-catenin增加,游离γ-catenin可与E-cad/β-catenin复合物中的β-catenin竞争,与E-cad形成复合物。从而使得游离β-catenin增加,导致β-catenin核易位和转录活性增加,进而导致侵袭相关基因表达,提高ESCC细胞侵袭能力[2]。此外,研究表明DSC2在ESCC侵袭和转移中有重要作用,它的缺失通过激活β-catenin途径启动肿瘤细胞转移,诱导上皮-间充质转化[25]。DSC2下调通过控制细胞-细胞附着和细胞骨架重排以及激活β-catenin信号传导的机制促进ESCC细胞侵袭[41-42]。提示DSC2的缺失与肿瘤分化程度低、局部淋巴结转移和预后差密切相关,其可能成为预测食管鳞状细胞癌患者预后的新分子标志物。
2.4 DSC2与其他肿瘤疾病DSCs是跨膜蛋白,属于桥粒钙黏蛋白家族,是桥粒的关键组成部分。在正常胃组织中DSCs的3种亚型(DSC 1~3)仅观察到DSC2。Ⅰ/Ⅱ期 胃 癌(Gastric cancer,GC)组 织 中DSC2的mRNA水平显著高于Ⅲ/Ⅳ期患者[43]。通过大肠杆菌氨苄青霉素分泌陷阱鉴定得知DSC2是胃癌上调的基因之一[43]。ANAMI K等[43]发现,DSC2在肠型GC中过表达,CDX2可以诱导DSC2的表达,表明DSC2的表达可能是肠型GC的关键调节因子。有研究[44]发现,胰腺导管腺癌中DSC2表达降低与患者生存期缩短、肿瘤分级增加和淋巴结阳性状态显著相关,表明DSC2在肿瘤进展和转移中起作用。研究发现,与人肝细胞系相比,肝癌细胞特别是在LM3细胞中,DSC2的表达显著下调,表明DSC2可能在肝癌的发展中发挥作用[24]。
3 小结与展望
多细胞生物的细胞间包含多种连接复合物,其中桥粒对保持组织完整性和维持生物体整体稳态至关重要。DSC2普遍存在于所有含有桥粒的组织中,是桥粒的组成部分。DSC2缺失或异常表达会降低细胞间黏附,破坏组织的完整性,表皮分化的误差调节,增加细胞增殖的倾向,侵入周围组织和转移,并导致某些疾病的发生发展。对于DSC2在某些疾病发展中作用的认识仍在不断深入,其可能作为一种癌基因,在肿瘤进展中起着至关重要的作用。研究人类不同疾病中DSC2的表达和调控机制的差异可为诊断和个体化治疗提供潜在靶点。DSC2在肿瘤进展中的作用需要进一步的研究来证明。
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29
广州大学学报(自然科学版)(2019年1期)2019-05-07
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
中成药(2017年8期)2017-11-22
天津科技大学学报(2016年1期)2016-02-28
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
中国现代医学杂志(2015年26期)2015-12-23
医学研究杂志(2015年7期)2015-06-22
西安交通大学学报(医学版)(2015年2期)2015-02-28
