
DFT法研究三聚甲醛开环聚合热力学
2024-05-08 13:27李增昌陆全华刘光臻
山东化工 2024年7期
李增昌,陆全华,刘光臻
(百色学院 化学与环境工程学院 广西城市水环境重点实验室,广西 百色 533000)
开环聚合是高分子化学中一个重要的领域,而环醚的开环聚合是开环聚合反应中具有代表性的一类。环醚开环聚合可用来合成多种应用广泛的产品,因此是研究较多的一大类反应。环状醚类化合物中,应用量最大的应该属于聚甲醛,聚甲醛英文缩写POM,是一种性能优良的热塑性工程塑料。其表面光滑有光泽,且其密度大,可在温度为-40~100 ℃的范围内长期使用。聚甲醛虽可由甲醛直接聚合,但由甲醛直接聚合得到的产品相对分子质量通常不高,并且需要较高的技术条件。据说当今只有少数大科技公司掌握了由甲醛直接聚合得到性能优良的聚甲醛。聚甲醛实际上更多的是由三聚甲醛即1,3,5-三氧六环开环聚合得到,或是与1,3-二氧五环共聚得到[1]。另外,它的耐磨性和自润滑性也比绝大多数工程塑料优越,且有良好的耐油性[2]。虽然聚甲醛也有不足,比如易降解、不耐酸碱等,但瑕不掩瑜,是用量仅次于聚酰胺和聚碳酸酯的第三大通用工程塑料,在很多场合得到了应用。
从环醚结构来看,单体的环张力是一个非常重要的热力学因素,这一因素决定了其能否进行开环聚合反应。其开环聚合的可能性,可根据热力学数据即反应前后的焓变或是自由能来进行判断。一般说来,开环的焓变主要来自环张力的释放。环张力大,焓变也就越大。自由能变化包括焓变和熵变两部分。而熵变一项与热焓相比数值较小,聚合趋势主要取决于焓变,而焓变又包括内能的变化和体积与压力的积两项。即ΔE和PΔV两项。在开环聚合过程中,PΔV的数值要远远小于ΔE的数值,故而内能的变化即ΔE基本上决定了开环的可行性。高分子化学的基本原理认为,开环聚合过程中的ΔE来自单体的环张力,而环张力主要是由相邻碳上直立键上的氢原子电子云之间斥力造成的[3]。一般情况下,六元环醚因可成椅式构象,像四氢吡喃。1,4-二氧六环等单体都是如此,张力较小,六元环状单体除1,3,5-三氧六环外,其他单体均不能发生聚合,正如前面所说,六元环状单体的内能变化主要来自椅式构象或是船式构象上直立氢原子之间的斥力,而多数情况下,这些氢原子的斥力较小,所以,该领域内的主要热点是研究三氧六环的聚合。
鉴于开环聚合在生产上的实际应用,系统地对开环聚合的热力学和动力学研究非常重要,然而这需要投入大量的人力和物力,即便如此,有些数据也难以获得。而理论计算,在某些方面可以弥补实际测量的不足。通过DFT法研究开环聚合热力学,可以为开发新型高分子材料提供理论指导。具体来说,研究开环聚合反应的热力学参数,如焓和熵,可以帮助优化聚合反应的条件,并预测聚合产物的性质。这对于合成新型功能高分子材料或改进现有材料的性能具有重要意义。因此,DFT法研究开环聚合热力学是一项具有潜在应用价值的研究工作。密度泛函理论的最早可以追溯到1920年前后,它的基本概念最早起源于托马斯和费米等人提出的Thomas-Fermi-Dirac模型[4],在1964年后由Kohn正式提出密度泛函理论,Kohn也因提出该理论获得1998年诺贝尔化学奖,这是对计算化学领域的一大肯定,也显示了理论在计算量子化学领域的核心作用。自1990年以来,DFT方法发展迅速,已经在理论计算的很多方面如键能、预测化合物结构和反应机理等方面取得巨大成功。这是一种应用广泛的第一性原理计算方法,密度泛函包含了电子相关能的计算,在提升了计算速度的同时,结果也能保持较高计算精度[20]。密度泛函理论是目前量子化学中最为流行和实用的计算方法之一,且其在材料科学、物理学、化学等领域都得到了广泛的应用。Guassian高斯量子化学计算软件是一个功能强大的工具,其有多种计算方法。其中的密度泛函理论,是目前备受青睐的计算方法,此计算方法具有计算精度高、计算速度快的优点,可以单纯通过计算来获取实验中无法获得中间体证据[5-8];能单独提出某些反应的机理,对反应现象和数据进行解释,或是对实验机理进行验证并能对实验数据做必要的补充[9-11]。本文就是利用高斯量子化学计算软件Guassian和Chem3D,对一系列开环单体开环聚合的热力学数据进行了计算,一方面可以用来弥补实际测量的不足,另一方面则可以回答高分子化学教学中的某些问题。
众所周知,环状单体的开环聚合,其动力主要来自环的张力,在许多高分子化学教科书中,均提到了1,3,5-三氧六环的结构与前者不同,但有什么不同,却均未给出答案。从教学的角度看,也有必要对1,3,5-三氧六环的结构与开环机理进行深入研究。
1 计算方法
本文全部通过密度泛函理论(Density Functional Theory),所有分子均在B3LYP水平上应用6-311G++(d,p)基组进行计算。选取了四氢吡喃、1,3-二氧六环、1,4-二氧六环和1,3,5-三氧六环(即三聚甲醛)四种六元环作对比。环状单体分别对椅式构象和船式构象进行优化和频率分析得到其稳定态的最低能量。环己烷、1,3-二氧六环、1,4-二氧六环三种环状化合物均是椅式构象能量较低,故取椅式构象的能量作为其单体的开环前单体的能量。1,3,5-三氧六环船式构象能量较低,故取其船式构象作为此单体的能量。由于Gaussian不适合对聚合物的片段进行直接计算,需用间接法求得。其中,环己烷开环产物的能量,由完全锯齿状正十二烷和完全锯齿状正己烷的能量差求得,1,3-二氧六环、1,4-二氧六环由锯齿状的含有端基的二聚体和含有端基的链状单体能量差求得。1,3,5-三氧六环的能量由带端基的螺旋形构象的二聚体和含有端基的单体之差求得。
2 结果与讨论
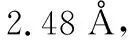
六元环及开链单元结构数据如表1所示。
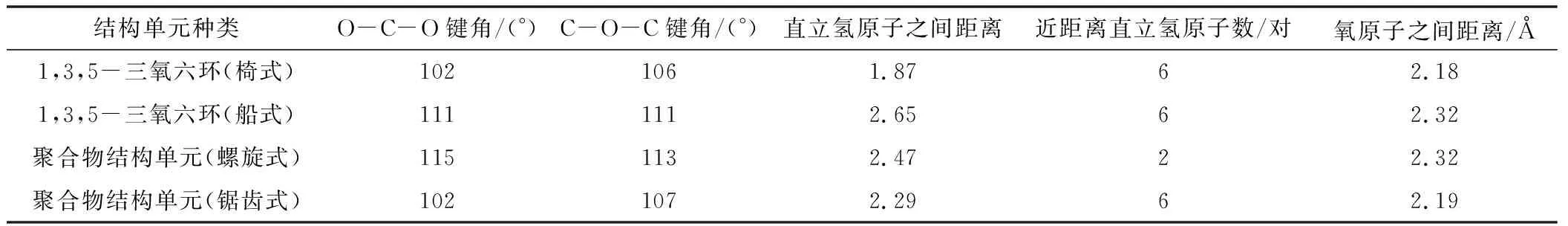
表1 六元环及开链单元结构数据
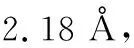
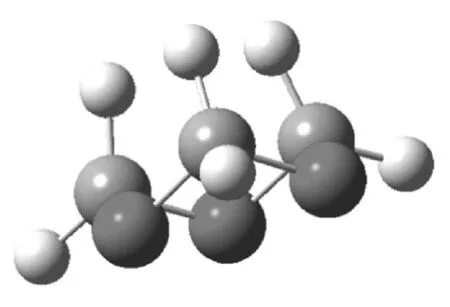
图1 1,3,5-三氧六环的椅式构象
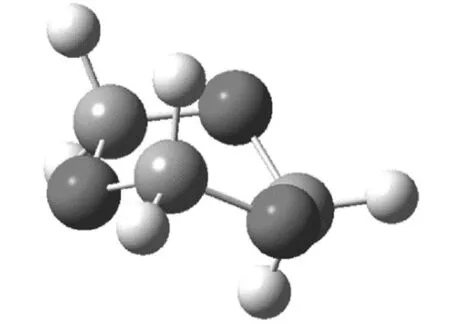
图2 1,3,5-三氧六环的船式构象
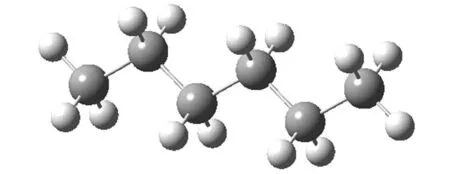
图3 正己烷构象
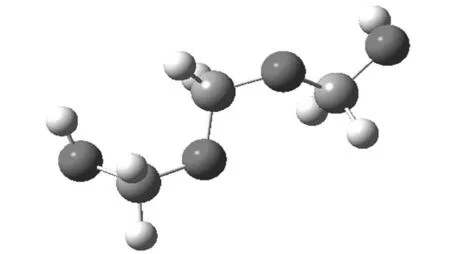
图4 聚甲醛结构单元构象图
各种六元环开环前后能量变化列于表2中,从表2可以看出,对于环己烷,其开环前的自由能更低,加上开环聚合过程是熵减少的过程,因此环己烷不可能发生开环聚合反应。四氢吡喃、1,4-二氧六环和1,3-二氧六环,有相似的情况,开环前后内能变化不大,考虑到聚合物熵的减少,自由能变化接近于零,小于理论计算的误差,因而判断为基本不能聚合,即使开环,也不能得到大分子量的聚合物。而1,3,5-三氧六环的自由能变化达到了-94 kJ·mol-1,因而是比较合适的开环聚合单体。
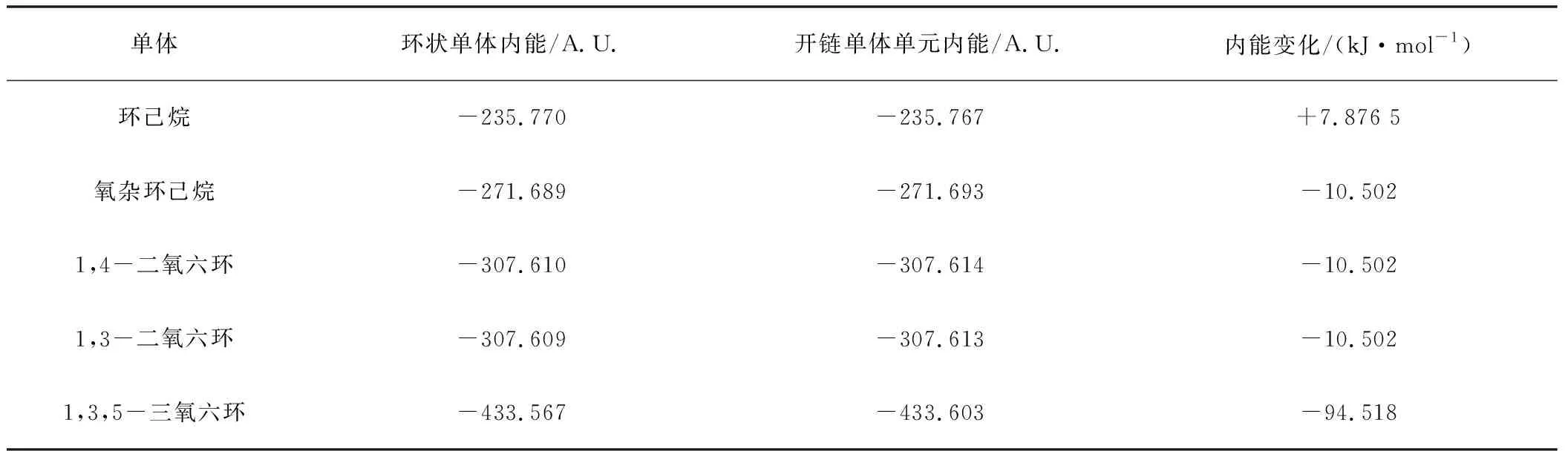
表2 六元环单体开环动力学数据
三氧六环开环的能量的变化,有几个方面的原因,从表1可知,一方面,船式构象与椅式构象相比,不论是C-O-C键角还是O-C-C的键角,都有所增加,结果是氧原子之间的距离变大,这减少了电负性较大的氧原子之间的斥力,另外因为,直立氢原子之间的距离也变大,这两方面的原因都使船式构象的能量有所降低,计算表明船式构象比椅式构象的能量要低86.6 kJ·mol-1。开链结构的聚甲醛,不是锯齿状结构,而是螺旋形结构,与船式构象的单体相比,一方面,尽管由于螺旋结构,氧原子距离没有明显比船式构象大,但不论是C-O-C键角还是O-C-C的键角,都有进一步增加, 这使得角张力大大减少,同时,直立氢原子的距离也大大地大于椅式构象中的氢原子的距离。尽管比船式构象中氢原子之间的距离稍小,但由于氢原子本身比较小,这个距离上其之间的斥力已经很小。同时,有斥力的直立氢原子的数量由六对减少为4对,使得螺旋形聚合物的能量较低。而锯齿状的聚合物结构中,一方面,C-O-C键角和O-C-C的键角,都比螺旋形结构的角度小,这使得角张力增大,同时,氧原子的距离也缩小了。另一方面,虽然氢原子之间的距离稍有增加,但是相斥的氢原子数量与螺旋形构想相比,则由2对变为6对。这两方面的原因都导致锯齿状构象的能量高于螺旋形构象。
3 结论
与环己烷相比,四氢吡喃、1,3-二氧六环、1,4-二氧六环四种结构相似,其分子的最低能量构象为椅式,而1,3,5-三氧六环分子的最低能量构象为船式构象。前面四种单体开环后的链式结构的最低能量构象为锯齿状构象,而聚甲醛的最低能量构象为螺旋形。在聚甲醛螺旋形构象中,由于C-O-C键角和O-C-C的键角增加导致角张力减少,同时氧原子距离较大,另外,相斥的直立键上的氢原子只有两对(其他结构中都是六对),这些因素都导致螺旋形构象的能量降低。计算表明,环己烷开环反应的自由能增加,因此环己烷通常不能发生开环聚合,四氢吡喃、1,3-二氧六环和1,4-二氧六环三种单体开环反应的自由能几乎为零,所以一般情况下,也不能发生开环聚合,而1,3,5-三氧六环开环聚合的自由能变化为-94.5 kJ·mol-1,可以在催化剂作用下,顺利进行聚合反应。
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29
现代塑料加工应用(2021年5期)2021-02-28
化工管理(2017年32期)2017-11-24
数理化解题研究(2017年16期)2017-07-21
绿色科技(2017年8期)2017-05-22
中学物理·高中(2016年8期)2016-08-08
中国塑料(2016年9期)2016-06-13
山西大同大学学报(自然科学版)(2016年6期)2016-01-30
中国塑料(2015年2期)2015-10-14
中国塑料(2015年9期)2015-10-14
