
先天性肝纤维化现状和研究进展
2024-05-08 13:43:54张一博李新华
临床内科杂志 2024年4期
张一博 李新华
先天性肝纤维化(CHF)是一种罕见肝病,该病的诊断目前主要是基于病理形态的一种临床综合征,其致病基因及临床表型均复杂多变,不论是在机制研究还是临床研究方面均存在不足。CHF病理生理学机制主要为胆管板畸形继发的胆道发育不良,胆管板重塑缺陷是其常见组织学特征。1856年,Parker在尸检报告中首次提及肝脏异常纤维化改变,1961年Kerr首次将其命名为“先天性肝纤维化”[1]。CHF发病率和患病率尚不明确,有文献统计患病率约为1/10 000~1/20 000[2],患者无明显性别差异。CHF临床表现多样但缺乏特异性,常合并多囊肾病及其他器官系统疾病,又因其罕见性,临床医师对其普遍认识不足,临床诊断率较低、误诊率高。目前针对该病尚缺乏有效的治疗手段,患者一旦进展到终末期肝病,病情无法逆转,因此提高认识,早发现、早治疗或可延缓病情的进展。近年来,随着超声、CT和MRI胰胆管成像(MRCP)等无创诊断技术及病理学的发展,CHF的临床诊断率正不断提高,正确认识该病的临床需求在逐步增加。国外大量研究结果显示纤维包囊蛋白/多囊蛋白(PKHD1)基因为该病最常见致病基因,我国的相关研究比较缺乏,据本院几十例患者基因检测的结果显示,大部分患者并不存在PKHD1基因的致病突变,因此探索新的致病基因及致病机制、发现新的治疗靶点是我国CHF研究亟需解决的问题。
一、CHF的致病机制
1.胆管发育异常:妊娠第8周左右,胎儿门静脉周围的原始肝母细胞像袖套一样包绕门静脉形成肝内胆管的胚胎前体——胆管板。第12周左右,胆管板逐渐开始重塑为各级胆管,胆管上皮细胞与门静脉间质组织的正常相互作用是诱导小胆管重塑的关键。第20周左右,肝内各级胆管完全成熟。不同的成熟停滞阶段可导致不同类型的疾病,一般认为晚期的胆管板成熟异常是CHF的发病机制之一,而中期的胆管板成熟异常更多导致先天性肝内胆管囊性扩张症(Caroli病)的发生,两种发育异常同时存在时可共同表现为Caroli综合征,但往往Caroli病的临床表现更为突出,以至于共存的CHF易被忽视。因此,所有疑似Caroli病的患者均需行肝脏组织病理活检,以确认是否存在CHF[2-3]。
门静脉高压是CHF的主要临床综合征之一,从胚胎学上讲,胆管的发育与肝脏血管的发育密切相关。门静脉海绵样变性(CTPV)是指当门静脉压力增高时,机体为减轻门静脉高压在门静脉周围形成侧支循环或阻塞后的再通,其病因尚未完全明确,主要可分为原发性和继发性。许多学者认为CTPV可出现在CHF的发病初期,是CHF的先天性组成部分,而不是随着疾病进程逐步进展所致[4]。
2.肝纤维化:CHF导致肝纤维化的病理生理机制尚不明确。肝星状细胞(HSC)活化被认为是肝纤维化形成的中心环节,HSC的活化主要由Kupffer细胞分泌的转化生长因子(TGF)-β1诱导。TGF-β1通过刺激成纤维细胞和相关细胞类型(包括肝内的HSC)分泌广泛的细胞外基质蛋白来介导其促纤维化作用[5]。同时,TGF-β1可促进胆管上皮细胞发生转化,起到类似于成纤维细胞的作用,推测这些转化细胞产生过多的细胞外基质分子也可能导致进行性肝纤维化[6]。既往研究也表明,TGF-β1可调节骨桥蛋白(OPN)过度表达,刺激纤维炎症的发生,参与CHF纤维化的进展[7]。
3.肾囊肿:CHF患者常伴有多囊肾改变,肾囊肿生成机制尚不清楚,有研究认为可能与PKHD1基因突变引起肾小管上皮纤毛功能障碍有关,但这无法完全解释多囊肾患者的病理改变。Cordido等[8]在动物模型中发现,从囊肿形成到不断进展这个复杂的过程中,可存在多种分子通路的改变,如细胞异常增殖信号通路[哺乳动物雷帕霉素靶蛋白(mTOR)、RAS-RAF-ERK、丝氨酸/苏氨酸激酶(AKT)等],蛋白激酶A(PKA)及腺苷酸环化酶6(AC6)调节的环磷酸腺苷(cAMP)生成等。
二、CHF相关致病基因
CHF的发病机制尚不明确,国外临床病例多见PKHD1基因突变。PKHD1位于染色体6p12,拥有至少86个外显子,是人类最大的基因之一。PKHD1基因最长的转录本由66个外显子(外显子2-67)组成,编码一个分子量为447 kD的蛋白——纤维囊蛋白/聚导管蛋白(FC/PD),该蛋白是一个大的类受体蛋白,大部分蛋白位于胞外,具有单个跨膜区和一个位于细胞质的尾巴。该蛋白主要位于胆管和肾小管上皮细胞的初级纤毛和顶端表面,可调节胆管上皮分泌胆汁及肾小管的代谢功能,PKHD1基因突变导致FC/PD蛋白异常,进而影响胆管及集合管发育,出现CHF及常染色体隐性遗传多囊肾病(ARPKD)表型[9]。PKHD1基因编码蛋白涉及纤毛发育异常,肝脏中存在多种具有初级纤毛的细胞类型,包括门静脉成纤维细胞(PF)和肝星状细胞(HSC),二者是肝损伤时引起纤维化的主要前体细胞类型[10]。
除PKHD1基因突变外,目前已知还有多种基因突变可导致CHF的病理改变:位于常染色体上的跨膜蛋白67(TMEM67)基因隐性突变可引起细胞纤毛生成受损、纤毛细长,在肝内导致胆汁排放受阻,引起进行性胆汁淤积伴γ-谷氨酰转肽酶(GGT)升高,最终患者肝脏病理可显示CHF[11-12]。ATP结合盒亚家族B成员4(ABCB4)基因突变引起多药耐药3(MDR3)蛋白缺陷通常引起遗传性胆汁淤积症,其组织学特征为胆管增生、门脉炎症和纤维化,但其肝脏病理特征亦可表现为CHF。卷曲螺旋及含2A的C2结构域蛋白(CC2D2A)基因突变相关的COACH综合征、编码肾囊素(NPHP)基因突变相关的肾小管实质性肾病-视神经盘缺损综合征(Senior-Loken综合征)及中心体和基底体相关蛋白(ALMS1)基因突变相关的Alstrom综合征均可表现出CHF的病理改变。随着研究不断进展,更多基因可能被发现,目前报道的CHF相关致病基因及临床表型总结见表1。
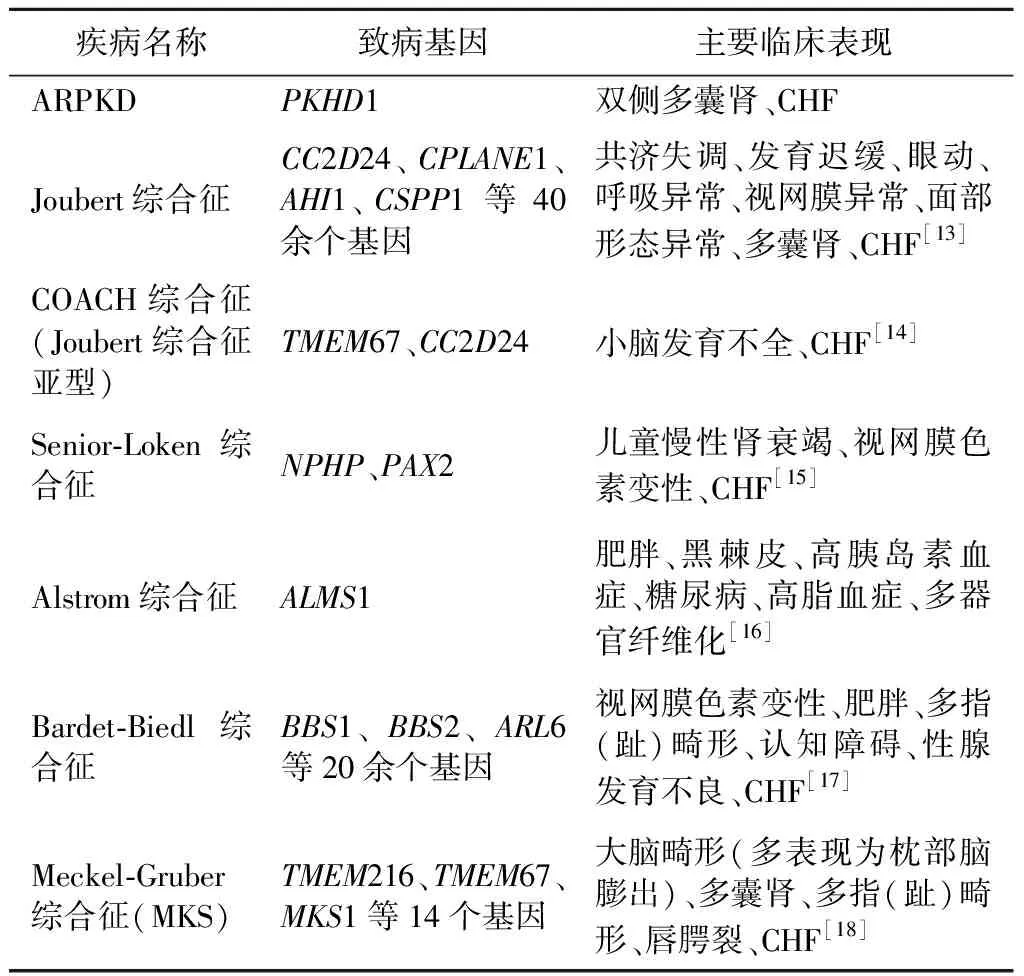
表1 CHF相关致病基因及临床表型
三、CHF的病理改变
CHF患者的肝脏大体标本或腔镜下可观察到肝脏萎缩,并且表面有大小不等的灰白色或灰黄色结节。显微镜下主要可观察到以下改变:(1)汇管区之间不同宽度的致密纤维间隔将肝实质分隔成肝岛,形成类似假小叶的结构——包含正常的脉管系统及完整的肝小叶,中央静脉位于中间;(2)胆管板重塑不良是该病的特征性病理改变,即胆管板畸形,表现为汇管区形态异常的未成熟小胆管——大量形态不规则的胆管增生或囊性扩张胆管结构;(3)肝内门静脉分支存在潜在异常或纤维化改变;(4)肝细胞坏死及变性不明显,少见炎细胞浸润。
在CHF合并胆管炎的患者中,病理切片可观察到胆汁淤积;在混合型患者中则同时具有上述两种病理特征。若CHF合并Caroli病,肝脏病理可观察到典型的交通性海绵状胆管扩张[19]。若合并多囊肾,可观察到患者双肾异常增大,肾实质见大量囊性结构,但肾小球数量一般正常;肾集合管增生、扩张、伸长呈放射状向肾门集中,从皮质蔓延至髓质,致皮髓质分界不清。
四、临床表现
CHF患者的临床表现复杂多变(可表现为肝脏、泌尿系统、中枢神经系统等异常),与肝脏相关的表型主要有发热、腹痛、黄疸等胆管炎表现及呕血、黑便、脾脏肿大(简称脾大)、脾功能亢进(简称脾亢)等门静脉高压表现。因此根据其临床表现,CHF可分为门脉高压型(最常见)、胆管炎型、混合型(兼具门静脉高压及胆管炎型表现)及隐匿型[20]。
国内外报道均显示门脉高压型CHF是最常见的类型,此类型患者以门静脉高压表现为主。儿童患者可早期出现明显临床症状,并常因反复呕血或黑便至医院首诊;成人患者则症状较轻或无明显症状,常因体检出现肝功能异常或其他疾病入院检查发现CHF,这表明不同年龄发病的CHF患者其临床表现可能存在差异。在脾亢患者中可观察到贫血和血小板减少,亦可见全血细胞减少[21]。胆管炎型CHF的主要表现为胆汁淤积和反复发作的胆管炎,此类患者可表现为发热和腹痛。混合型CHF患者合并有门静脉高压的表现和反复发作的胆管炎。隐匿型CHF一般无明显临床症状,患者在成年后常表现为不明原因的肝脾肿大,确诊主要依赖肝脏组织病理活检(简称肝活检)。
CHF可单独发生或与其他纤毛病合并发生,最常合并Caroli病和ARPKD。CHF患者合并Caroli病的临床表现取决于病变主要为肝内胆管扩张还是肝纤维化。前者临床可表现为胆管结石形成、胆管炎甚至肝脓肿,后者临床则主要表现为门静脉高压相关症状。约一半ARPKD患者在新生儿期出现肾脏增大、囊性变和肺发育不全[22],30%~50%的患病新生儿有严重的肺功能不全,甚者导致死亡。若ARPKD患者在新生儿期存活,则大多数最终将发展为系统性高血压,部分患者可有慢性肾功能不全,最终需要肾移植。在ARPKD患者中29%~68%的患者可表现为门静脉高压,临床上多囊肾患者应重视其肝脏检查,早期诊断及干预CHF[23]。
CHF其他可合并疾病还有Bardet-Biedl综合征[24]、Joubert综合征[25]、先天性糖基化异常(CDG)、Meckel-Gruber综合征[18]、常染色体显性遗传多囊肾病(ADPKD)等,临床应重视其肝外表现。
五、临床诊断
CHF的临床诊断主要依赖临床表现、影像、病理及基因检测的综合判断。任何不明原因门静脉高压患者临床均应考虑CHF的诊断。近年来随着门-窦血管病(PSVD)概念的提出[26],CHF是该类疾病中首先要鉴别的疾病。临床上出现门静脉高压相关临床症状突出而肝脏合成能力及排泄能力等功能基本正常或轻度异常时,应进行CHF及PSVD的排查。CHF门静脉高压型患者转氨酶轻度升高,在成人中转氨酶少有超过正常值上限(ULN)的3倍。临床上以胆管炎为主的患者可出现碱性磷酸酶(ALP)、GGT和胆红素明显升高,要注意与其他胆汁淤积性疾病相鉴别。
1.CHF的影像学诊断
放射学超声检查(US)是CHF诊断过程中的一线检查方法。CHF患者超声检查可见以下改变:肝叶比例欠协调(有患者表现为左叶异常肥大、右叶萎缩),肝实质回声增粗;肝内小胆管僵直扩张,可伴局灶性囊性或实性病变;肝门处胆管迂曲增厚;门静脉主干及左右支管腔内均可见低回声,周围见侧支循环(门静脉海绵样变性);肝动脉系统增宽等。合并ARPKD患者肾脏超声可见肿大的肾脏呈弥漫性高回声,皮质可表现为边缘较薄的低回声环,无法显示正常的皮髓质分界;随着疾病进展,肾小管不断扩张形成囊肿,肾实质内可见弥漫分布、大小不等的无回声区,肾实质内亦可出现大量的不显示声影的强回声灶[27]。
CT通过精确的体积测量和肝脏血管成像,相对US可更好地显示肝脏的大体形态,但是在诊断CHF方面的临床价值不如MRI肝脏成像(MR)并MRCP。CHF MR影像特点是胆管并非正常情况下向肝脏外周逐渐变细,相反,其在整个肝实质中扩张,常常在肝脏的边缘形成水池和小囊肿[28]。MRCP则可显示胆道树的异常分布——周围胆管轻度、均匀扩张,中央胆管相对细长——可发现US漏诊的病变,若出现明显的胆管扩张通常表明合并Caroli’s病。但MRCP也存在一定的局限性,其依赖T2加权序列将胆管内的液体显示为高信号结构,因此易受运动伪影的影响。有学者认为,半傅里叶采集单扫描涡轮棘波(HASTE)序列——一种超快的T2加权序列可彻底克服伪影问题,但由于其昂贵的价格在临床上并未取得广泛应用[29]。
2.CHF的病理学诊断
穿刺组织病理活检是诊断CHF的金标准,胆管板重塑不良为其特征性表现,若伴有汇管区粗大疏松纤维间隔而无典型假小叶形成,可高度提示CHF的诊断。CHF早期肝小叶通常正常,肝细胞形态也多为正常,有助于和肝硬化相鉴别,但在晚期CHF患者中,其病理改变与肝硬化难以区分,此时需结合病史、临床症状、实验室检查及影像学检查来综合诊断。
六、治疗
迄今为止,尚未有研究表明哪种药物治疗能够真正阻止甚至逆转CHF的病理过程,在动物实验中有一定疗效的抗肝纤维化的药物如秋水仙碱、血管紧张素Ⅱ受体阻断剂、干扰素-γ和吡非尼酮在临床中并未取得预期疗效[30]。CHF为胆管发育异常性疾病,对于胆管炎型CHF尤其伴有胆汁淤积的患者,熊去氧胆酸可能通过保护受损的胆管细胞免受胆汁酸的毒性作用、抑制肝细胞的凋亡从而在治疗中取得部分疗效[31]。临床上CHF患者的治疗主要为针对其并发症门静脉高压的处理,外科治疗主要为分流术和断流术,内科治疗主要包括内镜治疗食管胃底静脉曲张、口服β受体阻滞剂(如普萘洛尔、卡维地洛)降低门静脉高压等。肝移植是目前已知唯一治愈终末期CHF的方法[32],但供体和受体数量不平衡成为了目前肝移植面临的重要问题。因此,评估患者肝移植术后的风险与获益十分重要[33]。
七、总结及展望
CHF是一种罕见肝病,其相关基因的研究仍在不断发现中,临床上CHF除肝脏表现外,常伴发肾脏纤维及多囊肾病或其他多器官系统疾病表现。因此,在确定CHF的诊断后应注意排查其他器官系统,特别是肾脏或神经系统是否受累。肝活检在CHF的诊断和鉴别诊断中具有决定性价值,基因检测对于CHF的诊断及认识也有着重要意义[34]。目前对于CHF的治疗非常有限,亟需加强CHF致病机制及治疗靶点的研究,以期未来能有更多治疗手段来解决这类患者的病痛。
猜你喜欢
肝博士(2020年5期)2021-01-18 02:50:16
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:46
中国生物医学工程学报(2019年5期)2019-07-16 07:56:42
中国医学装备(2016年6期)2016-12-01 06:44:45
西南军医(2015年3期)2015-04-23 07:28:36
河南医学研究(2014年7期)2014-02-27 14:53:32
中华介入放射学电子杂志(2014年1期)2014-02-02 05:24:10
中国中西医结合外科杂志(2013年3期)2013-03-11 20:05:06
中国医学科学院学报(2011年1期)2011-03-25 13:58:43
河北医科大学学报(2011年11期)2011-03-25 10:17:21
