
Establishment of a prognosis predictive model for liver cancer based on expression of genes involved in the ubiquitin-proteasome pathway
2024-05-06 09:59HuaLiYiPoMaHaiLongWangCaiJuanTianYiXianGuoHongBoZhangXiaoMinLiuPengFeiLiu
Hua Li,Yi-Po Ma,Hai-Long Wang,Cai-Juan Tian,Yi-Xian Guo,Hong-Bo Zhang,Xiao-Min Liu,Peng-Fei Liu
Abstract BACKGROUND The ubiquitin-proteasome pathway (UPP) has been proven to play important roles in cancer.AIM To investigate the prognostic significance of genes involved in the UPP and develop a predictive model for liver cancer based on the expression of these genes.METHODS In this study,UPP-related E1,E2,E3,deubiquitylating enzyme,and proteasome gene sets were obtained from the Kyoto Encyclopedia of Genes and Genomes(KEGG) database,aiming to screen the prognostic genes using univariate and multivariate regression analysis and develop a prognosis predictive model based on the Cancer Genome Atlas liver cancer cases.RESULTS Five genes (including autophagy related 10,proteasome 20S subunit alpha 8,proteasome 20S subunit beta 2,ubiquitin specific peptidase 17 like family member 2,and ubiquitin specific peptidase 8) were proven significantly correlated with prognosis and used to develop a prognosis predictive model for liver cancer.Among training,validation,and Gene Expression Omnibus sets,the overall survival differed significantly between the high-risk and low-risk groups.The expression of the five genes was significantly associated with immunocyte infiltration,tumor stage,and postoperative recurrence.A total of 111 differentially expressed genes (DEGs) were identified between the high-risk and low-risk groups and they were enriched in 20 and 5 gene ontology and KEGG pathways.Cell division cycle 20,Kelch repeat and BTB domain containing 11,and DDB1 and CUL4 associated factor 4 like 2 were the DEGs in the E3 gene set that correlated with survival.CONCLUSION We have constructed a prognosis predictive model in patients with liver cancer,which contains five genes that associate with immunocyte infiltration,tumor stage,and postoperative recurrence.
Key Words: Liver cancer;Ubiquitin-proteasome pathway;Prognosis prediction;Gene expression;Immune infiltration
INTRODUCTION
The prevalence of liver cancer has been increasing,with an annual growth rate of up to 2%-3%[1] and survival rate of 18%in 2020[2].A total of 336400 new liver cancer cases were detected in China in 2016[3],and the sharply elevated incidence(18.0 per 100000) of liver cancer caused by sugar-sweetened food must be given extra attention[4].
Hepatitis B/C virus (HBV or HCV) infection,addiction to alcohol,liver cirrhosis,fatty hepatitis,and eating aflatoxin contaminated food are the risk factors for liver cancer[5].Imaging examinations for liver cancer include ultrasonography,dynamic contrast-enhanced computed tomography (CT),multimodal magnetic resonance imaging,18F-fluorodeoxyglucose positron emission tomography/CT,and so on.Virtual liver biopsy sampling pipeline for eliminating sampling bias may be the potential diagnostic method to investigate the nature of the lesions and etiology[6].In recent years,using statistical models combined with machine learning techniques to elevate the diagnostic accuracy of serum biomarkers such as α-fetoprotein and cell-free DNA or RNA has been widely applied to the early diagnosis of hepatocellular carcinoma[7].Additionally,surgical resection,transplantation,ablation,chemotherapy,and immunotherapy are common treatment options for liver cancer patients[8].However,effective surveillance and prediction of the prognosis of liver cancer still face multiple challenges due to the high heterogeneity of this malignancy.
The ubiquitin-proteasome pathway (UPP) is one of the key pathways of protein selective degradation in organisms[9],which is related to cell cycle,proliferation,differentiation,apoptosis,transcription,signal transduction,immune response,stress response,and extracellular effectors[10].The malfunction of the UPP is linked to various diseases,such as carcinogenesis,infection,autoimmunity,and inflammation.Based on The Cancer Genome Atlas (TCGA) datasets and 961 ubiquitin-proteasome system genes (UPSGs),Liuet al[11] found thatDDB1and CUL4 associated factor 13 (DCAF13),cell division cycle 20 (CDC20),and proteasome 20S subunit beta 5 (PSMB5) have excellent performance to predict the survival of liver cancer patients.Zhanget al[12] identified a seven-UPSG prognostic signature,of which autophagy related 10(ATG10) was found to participate in liver cancer development and prognosis through autophagy,immune response,and tumor metastasis.Therefore,proteasome inhibitors,as a class of potential and effective anti-tumor drugs,have attracted a growing body of attention from researchers.In this study,we examined the correlation of the expression of genes involved in the UPP with the prognosis of liver cancer,to screen out some key genes and construct a prognosis predictive model,in order to provide a new horizon for the role and potential mechanism of the UPP in the development of liver cancer.
MATERIALS AND METHODS
Gene sets and data collection
The UPP-related gene set included 857 genes from the UPP-related Kyoto Encyclopedia of Genes and Genomes (KEGG)pathways[13],among which 10 was related to E1,38 related to E2,651 to E3,112 to deubiquitylating enzyme (DUB),and 46 to the proteasome.
The expression data of 424 samples related to liver cancer were downloaded from The Cancer Genome Atlas (TCGA)database (https://portal.gdc.cancer.gov/).Three recurrent samples,50 normal tissue samples,and one sample without overall survival (OS) data were deleted and the remaining 370 samples were randomly divided into a training group (n=296) and a validation group (n=74) in a ratio of 4:1.Another validation set (GSE54236) was downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/).This data set included 162 samples,including 81 tumor samples.
Construction and validation of a prognosis predictive model
Univariate and multivariate regression analyses were performed to screen the prognostic genes in the E1,E2,DUB,and proteasome gene sets using the Survival (version 3.2-3) and Glmnet (version 4.0-2) packages in R.The threshold of univariate analysis wasP<0.1,and stepwise multivariate regression analysis was used to screen genes associated with OS.The risk score of the screened genes was calculated to construct a prognosis predictive model,and the prognostic ability was assessed using the receiver operating characteristic (ROC) curve drawn with Proc (version 1.16.2) package.According to the risk score,the patients were divided into either a high-risk or a low-risk group.The Maxstat (version 0.7-25) package in R was used to calculate the optimal cut-off value.The log-rank method was used to compare the difference in OS between the two groups,and the Survival (version 3.2-3) and Survminer (version 0.4.8) packages in R were used to draw the survival curve.In the validation group,the same method was used to verify the model.
Immunocyte infiltration
The abundance of 40 types of immune cells in each sample was analyzed using GVSA (version 1.32.0).The correlation analysis between the screened genes and immune-related indicators was performed using the Psych (version 2.0.8) and Corrplot (version 0.84) packages in R.
Analysis of correlation between clinical parameters and gene expression
The clinical parameters were compared between the high-risk and the low-risk groups using an independent sample ttest or two-sample Wilcoxon test,and Spearman correlation analysis was used to determine whether the gene expression and risk scores were statistically related to clinical parameters.The Psych (version 2.0.8) and Corrplot (version 0.84)packages in R were used for plotting.Univariate Cox regression analysis was used to determine the relationship between OS and clinical parameters,as well as the relationship between the gene expression and postoperative recurrence.
Identification of differentially expressed genes and enrichment analysis
The Limma package (version 3.40.2) was used to screen DEGs between the high-risk and low-risk groups,with the threshold set atP<0.05 and |log2FC| >1.Then the functional enrichment analysis was carried out using the database for annotation,visualization,and integrated discovery (DAVID,https://david.ncifcrf.gov/) to identify the enriched gene ontology (GO) terms and KEGG pathways of the DEGs.
Core DEGs in E3 gene set
As a specific substrate recognition element,E3 plays an important role in the ubiquitin-mediated proteolytic cascade[14].Because of its specificity,the relationship between the expression of genes in the E3 set and prognostic risk was analyzed separately.Similar to the screening method for DEGs,the Limma package was used to screen the DEGs in the E3 gene set between the high-risk and low-risk groups,and the screening threshold wasP<0.05 and |log2FC| >1.
Statistical analysis
IBM SPSS Statistics 21 and R (version 3.6.2) were used for statistical analyses.The Shapiro-Wilk test was used for normality test,and the independent samplettest or the two-sample Wilcoxon test were used to analyze the differences in variables between two groups.The chi-square test or Fisher's test was used for analysis of categorical variables.The logrank method was used to test the significance of survival data.
RESULTS
Construction of a prognosis-predicted model for liver cancer based on five genes
The clinical parameters of the whole samples,cases in the training group,and those in the validation group are listed in Table 1,with P value referring to the statistical results between the training group and the validation group.A total of five genes significantly related to prognosis were screened to construct a prognosis predictive model,includingATG10,proteasome 20S subunit alpha 8 (PSMA8),proteasome 20S subunit beta 2 (PSMB2),ubiquitin specific peptidase 17 like family member 2 (USP17L2),and ubiquitin specific peptidase 8 (USP8) (Table 2).In the training group,the area under the curve (AUC) values of the model for predicting 1-,3-,5-,and 10-year survival were 0.724,0.659,0.643,and 0.624,respectively (Figure 1A).All patients were classified into either a high-risk or low-risk group according to the score of risk.There was a significant difference in survival time between the high-risk and low-risk groups (P<0.001,Figure 1B).In the validation group,the AUC values of the model for predicting 1-,3-,5-,and 10-year survival were 0.614,0.66,0.64,and 0.649,respectively,and the low-risk group exhibited a higher survival probability than the high-risk group (P=0.012,Figure 1C and D),suggesting that the model can well predict the prognosis in liver cancer patients.In the GSE54236 data set,the AUC values of the model for predicting 1-and 3-year survival were 0.563 and 0.678,respectively,and the cases could be divided into a high-risk group and low-risk group by the risk score.There was a significantly difference in survival time between the high-risk group and low-risk group (P=0.014,Figure 1E and F).
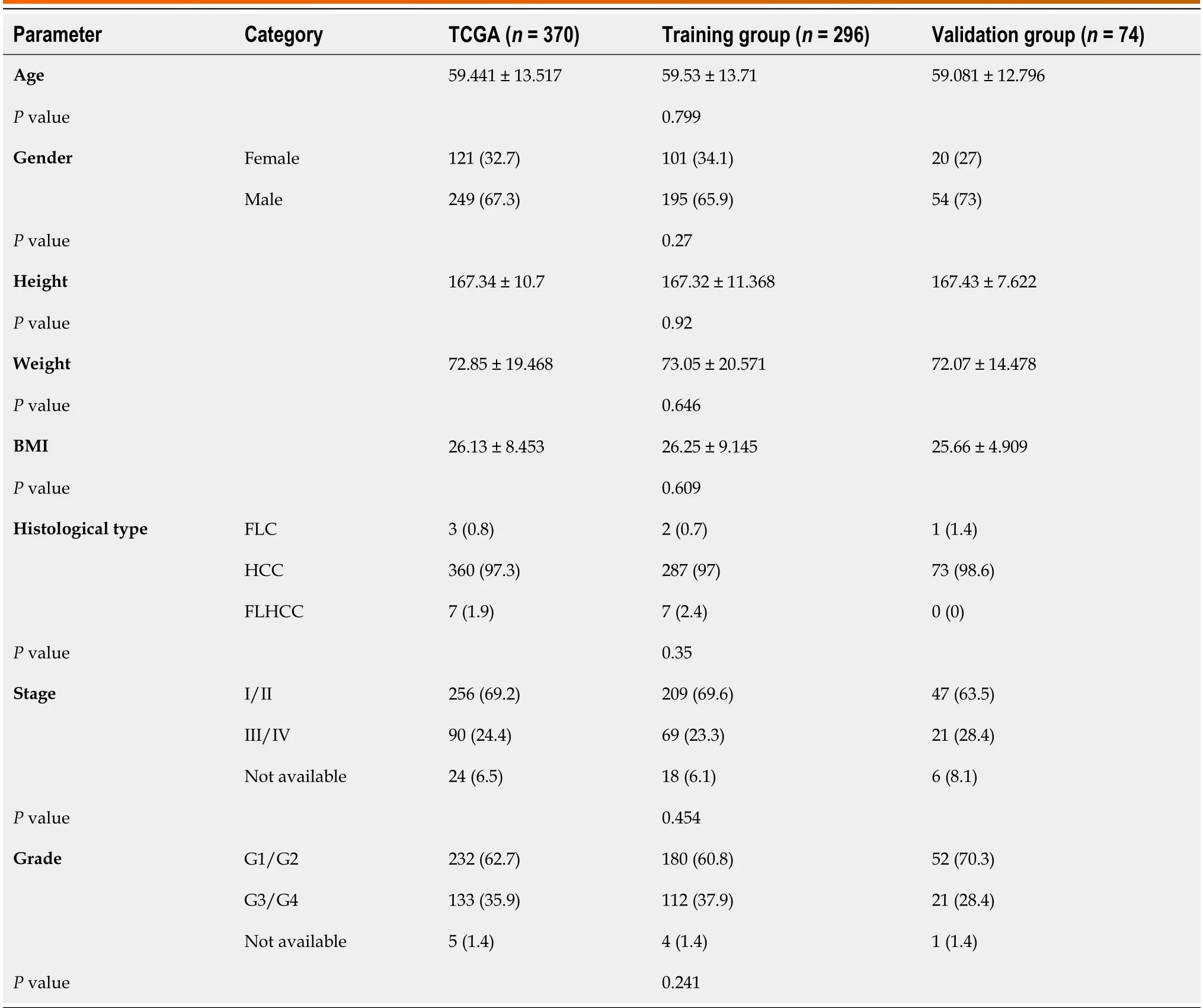
Table 1 Clinical parameters of the whole samples,cases in training group,and those in validation group,n (%)
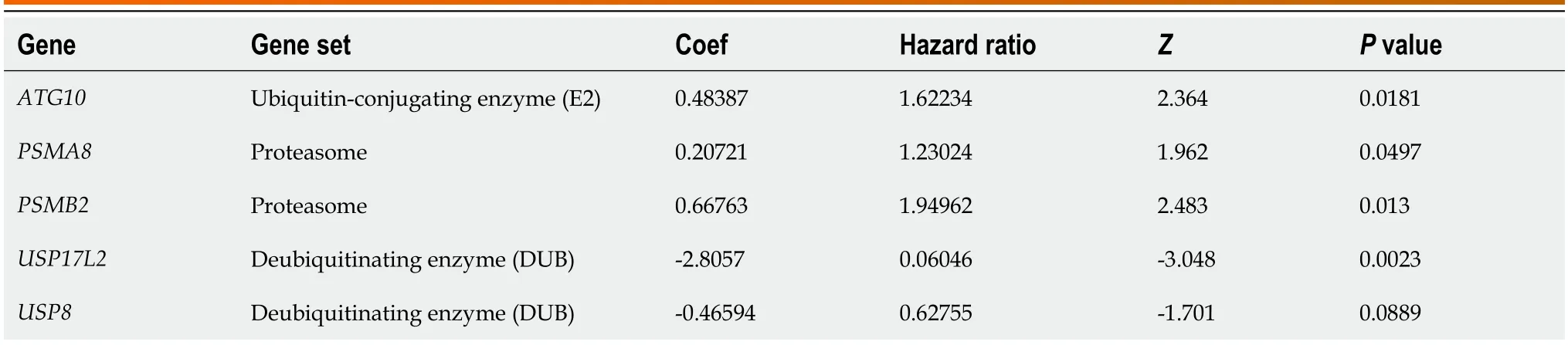
Table 2 The five genes significantly correlate with the prognosis of patients with liver cancer
Significance of expression of the five genes in immunocyte infiltration
Through the above analysis,we found that the expression levels of the five genes can predict the prognosis of liver cancer.Because immunocyte infiltration is commonly affected by gene expression,we then studied the correlation of the expression levels of the five genes with the abundance of 40 types of immune cells.PSMA8was associated with the abundance of the most immune cells,and the abundance of 28 immune cell types was significantly correlated withPSMA8expression levels.This was followed byUSP17L2,ATG10,USP8,andPSMB2,with 25,23,18,and 13 types of immune cells that were related to the expression levels of these genes,respectively (Figure 2).The abundance of most cells was negatively correlated with the expression levels ofATG10,USP17L2,andUSP8,whilePSMA8andPSMB2expression levels were positively correlated with the abundance of most cell types (Figure 2).

Figure 2 Immunocyte infiltration between cases with high and low expression levels of ATG10,PSMA8,PSMB2,USP17L2,and USP8.
The five genes are associated with tumor stages and postoperative recurrence
PSMA8andPSMB2expression and the risk score were significantly different between males and females (Figure 3A-C).For pathological and clinical stages,ATG10expression and the risk score were significantly different between T2 and T3 stages,PSMB2 andUSP17L2expression was significantly different between T1 and T3 stages (Figure 3D-G),the risk score was statistically lower in N0 stage than in NX stage (Figure 3H),and the expression levels ofATG10,PSMB2,andUSP17L2and the risk score were significantly different between stage I and stage II (Figure 3I-L).Moreover,PSMA8,PSMB2,USP17L2,andUSP8expression was all correlated with the upper limit of albumin results,among whichPSMA8andUSP17L2were positively correlated,andPSMB2andUSP8were negatively correlated with albumin results(Figure 3M).There was also a negative correlation between the risk score and the upper limit of albumin results,indicating that as the risk value increased,the albumin levels decreased,leading to an elevated prognosis risk for patients(Figure 3M).
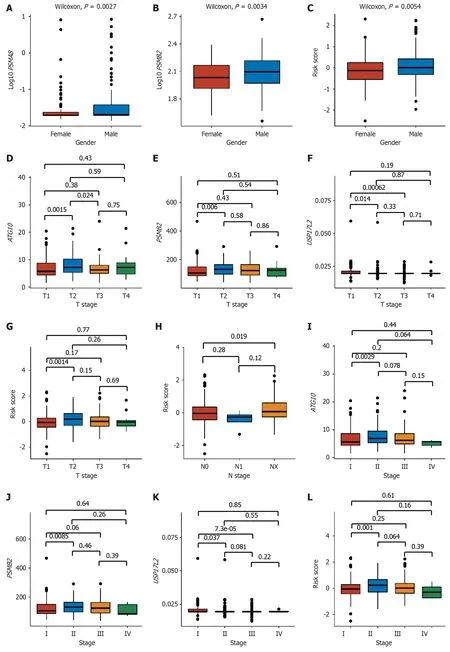
Figure 3 Correlation of expression levels of ATG10,PSMA8,PSMB2,USP17L2,and USP8 and risk score with clinical parameters. A-C:Gene expression and risk score between genders;D-G: Gene expression and risk score among T stages;H: Risk score among N stages;I-L: Gene expression and risk score among clinical stages;M: Correlation of gene expression levels and risk score with biochemical indexes;N: Correlation of gene expression levels and risk score with postoperative recurrence in liver cancer,with a hazard ratio (HR) >1 referring to a positive correlation and HR <1 referring to a negative correlation.P <0.05 indicated statistical significance.HR: Hazard ratio.
Postoperative recurrence included extrahepatic recurrence,local recurrence,intrahepatic recurrence,and new primary tumor.After univariate Cox analysis,ATG10,PSMA8,andUSP8,as well as the risk score,were found significantly correlated with postoperative recurrence (P<0.05,Figure 3N).
DEGs between high- and low-risk groups and their enriched pathways
A total of 111 DEGs were screened out between the high-risk group and low-risk group,among which 27 were upregulated and 84 down-regulated (Figure 4A).These DEGs were associated with 20 GO terms,comprising 9 biological processes,6 cellular components,and 5 molecular functions (Figure 4B).Five KEGG pathways enriched were GABAergic synapse,morphine addiction,neuroactive ligand-receptor interaction,retrograde endocannabinoid signaling,and cell cycle (Figure 4C).
DEGs in the E3 gene set between the high- and low-risk groups
Between the high-risk and low-risk groups,significant differences were observed in three genes within the E3 gene set:CDC20,Kelch repeat and BTB domain containing 11 (KBTBD11),and DDB1 and CUL4 associated factor 4 like 2 (DCAF4L2).In the high-risk group,CDC20andDCAF4L2exhibited elevated expression levels,whereasKBTBD11showed higher expression in the low-risk group.This suggested a negative correlation between the expression ofCDC20andDCAF4L2and survival,whileKBTBD11displayed a positive correlation with the prognosis of liver cancer (Figure 5).
DISCUSSION
The key factor to cell survival lies in the balance of protein synthesis and decomposition.The UPP is an ATP-dependent non-lysosomal protein degradation pathway,which is important for the body to regulate the level and function of intracellular proteins,thus efficiently and selectively degrading intracellular proteins.This study showed that the expression of the UPP genesATG10,PSMA8,PSMB2,USP17L2,andUSP8was significantly correlated with the prognosis of liver cancer.The prognosis model constructed based on these five genes could accurately predict the prognosis of patients (P<0.001 andP=0.012 in training and validation groups,respectively,Figure 1).These genes were statistically correlated with different clinical parameters and immune cell abundance (Figures 2 and 3).The model categorized all patients into either a high-risk group or a low-risk group,and a total of 111 DEGs were screened between the two groups,which were enriched in GO terms related to protein binding,GABA-A receptor,synapse,etc.,and KEGG pathways of retrograde endocannabinoid signaling,neuroactive ligand-receptor interaction,morphine addiction,GABAergic synapse,and cell cycle (Figure 4).
Those five genes were found to promote the development of many malignant tumors,including liver cancer[15-22].Our results showed that the increased expression ofATG10,PSMA8,andPSMB2increased the risk of death (P=0.018,0.049,and 0.013,respectively),while the increased expression ofUSP17L2andUSP8decreased the risk of death (P=0.002 and 0.089,respectively).According to previous studies,the overexpression of ATG10 and PSMB2 in tumors promoted the invasion or metastasis of tumor cells[16,18],and USP8 showed the opposite effect[21,22].Besides,PSMA8 could affect the progression and prognosis of colorectal cancer due to its strong association with PSMB2[23].Interestingly,higher PSMA8 expression levels were correlated with good prognoses for breast cancer through epigenetic regulation[24].In our study,it was found thatPSMA8was positively correlated with the prognosis of patients with liver cancer.On the contrary,USP17L2 has been found to be overexpressed in a variety of tumors[19,20],which is similar to our results.However,recent studies have found that up-regulation of USP17L2 causes chemotherapy resistance in colorectal cancer,and knockdown of USP17L2 could overcome bromodomain and extra-terminal domain inhibitor resistance in prostate cancer cells[25,26].Hence,the role of USP17L2 in liver cancer still requires further exploration.
The global immune system functions pose great technical challenges to the research of tumor-immune interaction[27,28].Because immune infiltration plays a key role in the development of liver cancer[27],we conducted a thorough correlation analysis to identify the immune cell types associated with the prognosis model.Minor alterations in the distribution of immune cells could potentially exert diverse impacts on the progression of tumors[29].In this study,myeloid dendritic cells were the immune cell type with a significant difference in abundance only between groups with high and low expression of thePSMB2gene,as well as neutrophils and Th17 cells between groups with different expression of theUSP17L2gene (Figure 2).However,no significant correlation was found between tumor-infiltrating immune cells and gene expression,and it is imperative to conduct additional confirmation and validation in an independent cohort.Furthermore,exploring the connection between the expression levels of some checkpoints and immune infiltration,as well as the tumor microenvironment,will be a hotspot for future research.
Moreover,the expression of one or more of the five genes and the risk score were different among different T,N,and clinical stages (Figure 3).It is widely known that tumor stage is a key prognostic factor for malignant tumors[30].In addition,all genes exceptATG10and the risk score were correlated with the upper limit of albumin results (Figure 3).The risk score was not only statistically significant in different stages,but also negatively correlated with the upper limit of albumin results and postoperative recurrence,which proves that the model developed in this study has appreciated value in clinical prediction of recurrence and prognosis.
E3 is the key factor in the UPP,which can specifically recognize different substrates and show high selectivity in protein degradation.Therefore,we analyzed the E3 gene set independently of E1,E2,DUB,and proteasome-related genes.Finally,the expression levels ofCDC20,KBTBD11,andDCAF4L2were identified as significantly different between the high-risk and low-risk groups,which were also included in the above 111 DEGs.CDC20 plays a vital role in chromosome segregation and mitosis[31].It regulates the stability of phosphorylated mitotic centromere-associated kinesin in metaphase-anaphase transition[32],which may play a role as a cancer protein to promote the development and progression of liver cancer.In the study of Zhenget al[33],CDC20,proliferating cell nuclear antigen,and minichromosome maintenance complex component 6 synergistically affect the regulation of the cell cycle and may be potential prognostic factors for liver cancer.Shiet al[34] found that CDC20 serves as a crucial factor in the development of hepatocellular carcinoma (HCC) by controlling the prolyl-4-hydroxylase domain 3 protein.By analyzing four expression profiles from the GEO database,it was found that the up-regulation of CDC20 in HCC tissues indicates poor OS and disease-free survival[35].Recently,KBTBD11was identified as a newly discovered adipogenesis-related gene[36].In diverse cancer types,such as colorectal cancer,HCC,and head and neck squamous cell carcinoma,the expression of KBTBD11 was significantly decreased in tumor tissues as compared to normal tissues[37].This is consistent with our result that patients in the high-risk group had lowerKBTBD11gene expression levels.DCAF4L2 is a member of the E3 complex,which is usually used as a mediator of protein-protein interaction and negatively regulates NF-κB signal transduction.Overexpression of DCAF4L2 has been observed in human colon cancer[38].In a study of HCC,overexpression of DCAF4L2 is a common feature of nonalcoholic steatohepatitis-associated HCC and viral hepatitis-associated HCC,which can be used as a candidate therapeutic target for HCC[39].We also found overexpression ofDCAF4L2in high-risk patients,which suggested a poor prognosis in patients with liver cancer.
One of the main shortcomings of this study is the lack of clinical cases.All the data were from TCGA and GEO,resulting in the lack of clinical data for some patients,and it was unable to validate the expression of the five genes and comprehensively analyze their correlation with clinical and prognostic indicators.This is a preliminary study,and the results reported are exploratory.We intend to validate these results and the detailed mechanisms in future studies.
CONCLUSION
In conclusion,we have used gene expression data in TCGA to screen genes involved in the UPP pathway that significantly correlate with the prognosis of liver cancer.Our findings indicate that the UPP plays an important role in the development of liver cancer,which provides new insights into the early prediction of prognosis and precision medicine in liver cancer.
ARTICLE HIGHLIGHTS
Research background
The ubiquitin-proteasome pathway (UPP) is crucial for selective protein degradation,and its dysfunction is linked to various diseases,including cancer.Proteasome inhibitors are emerging as potential anti-tumor drugs.This study explored the association between UPP gene expression and liver cancer prognosis,aiming to identify key genes and develop a predictive model.By doing so,the research seeks to offer novel insights into the role and potential mechanisms of the UPP in liver cancer development,contributing to the ongoing exploration of effective therapeutic strategies for liver cancer.
Research motivation
Due to the high tumor heterogeneity,effective surveillance and predication of the prognosis of liver cancer still face multiple challenges.This study was performed to analyze the relationship between the expression of genes in the UPP and the prognosis of liver cancer and construct a prognosis predictive model for this malignancy.
Research objectives
The study aimed to investigate the prognostic significance of genes in the UPP in liver cancer.Using gene expression data from The Cancer Genome Atlas (TCGA) and gene expression comprehensive (GEO) databases,the study identified key genes involved in the UPP,constructed a prognostic predictive model for liver cancer,and explored the associations of the model with immune cell infiltration and clinical parameters,in order to enhance liver cancer prognosis prediction and provide insights into the role and potential mechanisms of the UPP in liver cancer development,contributing valuable information for precision medicine in the context of liver cancer management.
Research methods
The research employed diverse methodologies,utilizing UPP-related gene sets and patient data from TCGA and GEO databases.A prognostic model was constructed using univariate and multivariate regression analyses,involving five key genes (ATG10,PSMA8,PSMB2,USP17L2,and USP8).The model demonstrated robust predictive abilities for liver cancer prognosis.Immunocyte infiltration analysis and correlation studies with clinical parameters provided additional insights.Differentially expressed genes and enrichment analyses shed light on relevant pathways.The study's comprehensive approach contributes a nuanced understanding of UPP gene implications in liver cancer prognosis.
Research results
This study investigated the role of the UPP in liver cancer,identifying five key genes (ATG10,PSMA8,PSMB2,USP17L2,andUSP8) associated with prognosis.A predictive model was constructed and validated using TCGA and GEO datasets.The study highlighted differential gene expression between the high-and low-risk groups and enriched relevant pathways.Additionally,differentially expressed genes in the E3 gene set (CDC20,KBTBD11,andDCAF4L2) were identified as significant.The findings provide valuable insights into liver cancer prognosis,immunology,and potential therapeutic targets.
Research conclusions
We have used gene expression data in TCGA to screen genes in the UPP that significantly correlated with the prognosis of liver cancer.Our findings indicate that the UPP plays an important role in the development of liver cancer,which provides new insights into the early prediction of prognosis and precision medicine in liver cancer.
Research perspectives
This is a preliminary study,and the results reported are exploratory.We intend to validate these results and the detailed mechanisms in future studies.
FOOTNOTES
Co-first authors:Hua Li and Yi-Po Ma.
Co-corresponding authors:Xiao-Min Liu and Peng-Fei Liu.
Author contributions:Liu XM and Liu PF conceptualized and designed the research;Li H and Ma YP collected the data and wrote the manuscript;Wang HL conducted the data mining and prepared the figures;Tian CJ,Guo YX,and Zhang HB conducted the bioinformatics analysis;all authors were involved in the critical review of the results and have contributed to,read,and approved the final manuscript.Li H and Ma YP contributed equally to this work and are the co-first authors.Liu XM and Liu PF contributed equally to this study and are the co-corresponding authors.There are two primary reasons behind appointing Li H and Ma YP as co-first authors,and Liu XM and Liu PF as co-corresponding authors.First,our research was conducted through a collaborative effort,and the selection of first and corresponding authors aptly mirrors the distribution of responsibilities and the shared commitment of time and effort needed to carry out the study and produce the resulting paper.This approach ensures effective communication and facilitates the management of post-submission matters,ultimately enhancing the paper's overall quality and reliability.Second,each of these researchers made substantial and equal contributions throughout the entire research process.Designating them as co-first authors or cocorresponding authors not only acknowledges and respects their equivalent input but also highlights the spirit of teamwork and collaboration that characterized this study.In summary,the choice to designate Li H and Ma YP as co-first authors,and Liu XM and Liu PF as co-corresponding authors is appropriate for our manuscript as it accurately reflects our team's collaborative ethos and equal contributions.
Supported bythe Tianjin Municipal Natural Science Foundation,No.21JCYBJC01110.
Institutional review board statement:TCGA is a public database.The patients involved in the database have obtained ethical approval.Users can download relevant data for free for research and publish relevant articles.Our study was based on open-source data,so there are no statements on ethics approval and consent.
Informed consent statement:Our study is based on open-source data,so there are no statements on informed consent.
Conflict-of-interest statement:All authors declare that they have no competing interests to disclose.
Data sharing statement:Publicly available datasets were analyzed in this study,and these can be found in the TCGA database (http://portal.gdc.cancer.gov/).
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Hua Li 0000-0001-5257-889X;Xiao-Min Liu 0000-0002-7533-3809;Peng-Fei Liu 0000-0002-2971-3800.
S-Editor:Liu JH
L-Editor:Wang TQ
P-Editor:Zheng XM
 World Journal of Clinical Oncology2024年3期
World Journal of Clinical Oncology2024年3期
- World Journal of Clinical Oncology的其它文章
- Current interventional options for palliative care for patients with advanced-stage cholangiocarcinoma
- Ferroptosis biomarkers predict tumor mutation burden's impact on prognosis in HER2-positive breast cancer
- Clinical application of reserved gastric tube in neuroendoscopic endonasal surgery for pituitary tumor
- Nomogram based on multimodal magnetic resonance combined with B7-H3mRNA for preoperative lymph node prediction in esophagus cancer
- Transarterial chemoembolization plus stent placement for hepatocellular carcinoma with main portal vein tumor thrombosis: A meta-analysis
- PD-1 antibody in combination with chemotherapy for the treatment of SMARCA4-deficient advanced undifferentiated carcinoma of the duodenum: Two case reports