
肯尼迪病3例临床特征分析并文献复习
2024-04-29 12:06柏亚真张登科杜敢琴富奇志
中风与神经疾病杂志 2024年4期
柏亚真, 张登科, 杜敢琴, 富奇志
肯尼迪病(Kennedy's disease,KD),又称脊髓延髓肌萎缩症(spinal and bulbar muscular atrophy,SBMA),是一种罕见的进行性神经退行性疾病,由X染色体上雄激素受体(androgen receptor,AR)基因的第一个外显子中CAG 重复序列的异常扩增引起[1],发病率为1/40 000~1/100 000[2]。KD 的核心症状为肌无力,大多伴有雄激素不敏感表现。少数不典型病例可伴有自主神经功能障碍、认知功能障碍等症状[3]。肯尼迪病少见,且临床表现多样,需要与肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)、脊髓性肌营养不良症(spinal muscle atrophy,SMA)等多种疾病相鉴别,早期误诊率极高[2]。现报道3例就诊于河南科技大学第一附属医院神经内科的KD患者,并结合相关文献,进行临床特点分析,以提高对KD 的识别与诊断。
1 病例资料
例1,男,60岁。以“进行性四肢无力14年,加重2 月”为主诉入院。患者于14 年前开始出现右下肢无力,主要表现为间歇跛行,未在意,症状缓慢进展,并逐渐出现左下肢无力;5 年前开始出现上楼梯困难,行肌电图等相关检查(具体不详),未特殊诊治;2年前出现双上肢平举时震颤;1年前出现蹲下后起立困难,易跌倒;4月前出现双上肢沉重、乏力,握筷、洗头费力;近2 月来上述症状进行性加重明显,只能行走50 米;病程中无晨轻暮重、肢体麻木、明显肌肉萎缩及肥大、肌肉疼痛、“肉跳”。既往“冠心病”病史,无阳性家族史。体格检查:乳房女性化,阴茎短小;舌肌萎缩、束颤;四肢肌张力减低,双上肢肌力Ⅳ+级,双下肢肌力Ⅳ级,四肢腱反射消失,病理征(-);双上肢平举时可见姿势性震颤;感觉(-)。辅助检查:肌酸激酶446 U/L ↑,促黄体生成素(LH)13.4 mIU/ml↑,风湿免疫相关、甲状腺功能、肝肾功能均正常。肌电图示神经源性损害;颈椎MR:颈椎退行性病变,C3/C4、C4/C5、C5/C6、C6/C7椎间盘轻度突出。腰椎MR:腰1/2、2/3、8/4、4/5、5/骶1 椎间盘膨出;基因检测:AR基因CAG重复次数为48次。
例2,男,70 岁。主因“面肌抽动、口角歪斜27年,四肢无力进行性加重7年”来诊。患者于27年前出现面部肌肉抽动,伴口角歪斜,未治疗;7年前出现双下肢无力,活动后易疲劳,后逐渐出现双上肢无力,伴四肢肌肉萎缩、言语不清,偶有饮水呛咳,双上肢夜间偶有麻木;2 年前出现下蹲后站立困难,爬楼梯困难;病程中无肌肉疼痛、痉挛及“肉跳”。既往“高血压、脑梗死”病史,无阳性家族史。体格检查:双侧乳房女性发育,构音障碍,左侧额纹较右侧变浅,左眼闭合无力,右侧鼻唇沟浅,双侧鼓腮差,左侧明显,伸舌居中,舌肌萎缩伴纤颤,四肢肌力Ⅳ级,四肢肌肉萎缩,四肢腱反射消失,双上肢腕关节以下及双下肢膝关节以下痛觉过敏;病理征(-)。辅助检查:肌酸激酶(CK) 517 U/L↑,促黄体生成素(LH)14.3 mIU/ml↑,风湿免疫相关、甲状腺功能、肝肾功能均正常。肌电图示广泛神经源性损害;颈椎、左大腿、左小腿MR:C4/C5、C5/C6、C6/C7 椎间盘稍向后膨出;左侧大腿、小腿平扫未见明显异常;基因检测:AR基因CAG重复次数为42次(见图1)。

图1 患者2的基因检测毛细血管电泳图
例3,男,57岁。主因“双下肢无力12年,双上肢无力5 年,加重1 周”来诊。患者于12 年前出现双下肢无力,表现为活动后无力,休息后可缓解,无晨轻暮重、双眼睑下垂等症状,未在意;5年前患者出现双上肢无力,活动后双上肢无法抬举,双手无法伸展,未治疗;1 周前自觉上述症状加重,上楼梯、洗头困难,休息后缓解不明显;病程中无四肢麻木、肌肉疼痛、痉挛及“肉跳”。既往“冠心病”病史,无阳性家族史。体格检查:双侧乳房女性发育;构音障碍,可见舌肌萎缩、纤颤及下颌震颤(见图2)。双手大、小鱼际肌萎缩,四肢肌张力正常,肌力Ⅳ级,四肢肌肉未见萎缩,腱反射正常,感觉及病理征(-)。辅助检查:肌酸激酶(CK) 498 U/L↑,促黄体生成素(LH)14.4 mIU/ml↑,风湿免疫相关、甲状腺功能、肾功能均正常,肝功轻度异常。肌电图示广泛神经源性损害(见图3);左上臂、左大腿MR:左上臂平扫未见异常;双侧大腿软组织信号改变,考虑脂肪化可能;基因检测:AR基因CAG重复次数为44次。本研究病例的临床表现见表1,实验室检查见表2,辅助检查见表3。

表1 3例肯尼迪病患者的临床表现
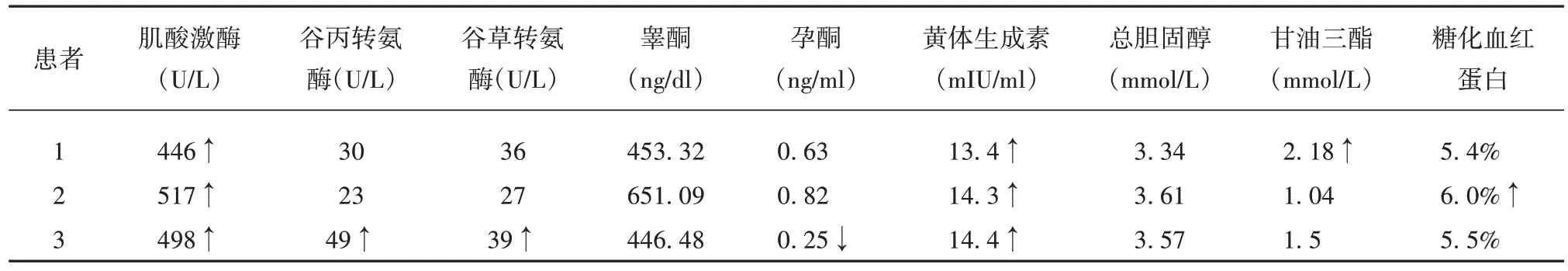
表2 3例肯尼迪病患者的实验室检查

表3 3例肯尼迪病患者的辅助检查

图2 患者3的阳性体征

图3 患者3的神经传导速度曲线
2 讨 论
KD 是Kennedy 于1968 年在描述来自两个不同家族的11 例患者中首次提到,并注意到伴性隐性遗传模式[4]。1991年,La Spada等[1]确定此病由编码谷氨酰胺的雄激素受体(AR)基因中扩增的三核苷酸重复序列引起的。已经证实突变AR 蛋白的毒性是配体依赖性的,换句话说,肯尼迪病是一种雄激素依赖性疾病[5]。
肯尼迪病通常始于中年,范围为18~64 岁,慢性病程[6]。在本研究中,3例患者出现首次症状的年龄一致,约为45 岁左右,病程持续数十年,这与先前报道一致。除患者2 外,患者1 和患者3 首次就诊在症状出现数年后,且3 例患者首次就诊时间距离确诊KD 短则7 年,长则27 年。韩国2023 年最新发表的一项包含157 例KD 患者的横断面研究[7]表明,患者平均病程为(11.6±7.0)年,在症状出现后约5.8 年才进行首次医学评估,确诊KD 还需要0.8 年,与其他国家的研究结果基本一致,表明临床医师早期识别KD并不容易。先前有研究报道,诊断患有肌萎缩侧索硬化(ALS)的患者实际上有2%合并有KD[8]。初始症状较轻且疾病发展缓慢可能是患者未及时就诊的原因,而首诊往往未能确诊则可能由于KD临床表现异质性强,临床医师对疾病认识不足,从而造成漏诊误诊。
KD 的主要临床表现包括延髓和四肢肌肉无力、束颤、震颤、痉挛、感觉障碍、男性乳房发育以及性功能障碍,初始症状在不同国家研究中有所差别,但大多数国家研究表明下肢无力为最常见的初始症状[7,9-11]。本研究评估了3例患者的核心症状,患者1和3 均为下肢无力起病,而患者2 以面部肌肉抽动、口角歪斜起病,这在以往的报道中并不常见,早期极易被误诊为面神经炎或单纯的面肌痉挛。球部受累也是KD 的核心症状之一,3 例患者均存在构音障碍,且患者均有轻至中度的舌肌萎缩与束颤,患者2还存在饮水呛咳,这进一步体现了构音障碍或吞咽困难以及舌肌萎缩是KD 具有标志性意义的体征。此外,患者3 出现下颌的不自主抖动,又被称为“颤抖下巴”,可能由于口周肌束震颤引起。男性乳房发育为KD 最常见的非神经系统症状,Spereld 等[12]对34 例德国患者的研究中,最初表现为男性乳房发育症的患者占52%,肌肉无力仅占4%。本文中,3例患者均出现不同程度的乳房增大,目前认为是雄激素不敏感所致,大多数患者还会出现不孕、性功能障碍等表现,可作为临床诊断KD的一个重要线索。在疾病的后期,超过一半的患者可能出现远端肢体的感觉症状,如麻木和刺痛[13]。本文3例患者中只有患者2出现感觉症状,这可能与其长达27年的病程有关。
既往实验室检查显示,大多数患者的血清肌酸激酶(CK)水平升高[14],一些患者还患有高脂血症、肝功能异常和葡萄糖耐量异常[15]。本文中3 例患者均有CK 升高,患者3 肝酶轻度升高,提示轻度肝功能异常;患者2 糖化血红蛋白稍高,患者1 出现甘油三酯异常,这提示KD 的代谢紊乱存在异质性,可能与多种因素有关。激素水平异常也是KD 常见的表现,如雌激素水平增高,睾酮水平异常。德国最近的一项研究调查[16]显示分别有22.5%和12.5%的KD患者出现黄体生成素(luteinizing hormone,LH)和卵泡刺激素(follicle stimulating hormone,FSH)升高。该研究还发现20.3%的KD 患者的睾酮水平升高。Cho等[7]的研究显示了类似的结果,证实了在来自不同国家的KD的大量患者中观察到的这种激素特征。与既往报道一致,本文3 例患者均有LH 的升高,但FSH 及睾酮均未观察到类似结果,这可能与患者例数过少有关。
肌电图及电生理检查是诊断KD的重要手段,超过90%的KD 患者出现异常的神经传导。本报道中的3 例患者肌电图检查均提示神经源性损害,电生理提示运动和感觉神经均受累,表现为不同程度传导速度减慢、波幅降低或消失、潜伏期延长及F 波引出率下降,与国内外报道相符合[6,17]。据报道,一部分患者可拥有正常的运动神经传导,提示感觉神经受累较为明显是KD 的特征性表现[6]。值得关注的是,虽然3 例患者的神经电生理检查均有明显的感觉神经传导异常,但只有1 例出现肢体麻木症状。这种现象在先前研究中已被多次提及,且被认为可能与AR 基因在感觉神经轴索高表达有关[18]。同时,通过神经电生理检查可有效地与ALS 相鉴别,ALS 通常合并上运动神经元症状且不累及感觉神经[2]。
目前基因检测仍为KD诊断的金标准,正常受试者AR 基因外显子中的CAG 重复数约为13~30,但在KD患者中,外显子是其正常长度的2.3倍,欧盟神经科学最新(EFNS)指南以CAG 重复数≥35 为诊断依据[19],但文献报道中通常认为CAG重复长度范围>38是致病的。CAG重复次数与KD进展速度及血清CK水平无关,但与发病年龄呈负相关,即CAG 重复次数越高,发病年龄越早,这在多个国家的先前研究中已被报道[7,9,11]。本文报道病例中,3 例患者CAG 重复次数均>38,未观察到与年龄明显的相关性,需要增加病例数进一步研究。先前有研究报告称,CAG重复次数越高,患者的运动神经症状越显著,重复次数低,则感觉神经症状显著[20]。Cho等[7]研究再次显示了类似的结果,表明CAG 重复长度与感觉神经异常有关。这些感觉主导电生理参数可作为鉴别KD与其他运动神经元病的替代标志物。
综上,本文报道了3 例KD 患者的临床特征及辅助检查等,并结合相关文献分析,为临床医师对该病的进一步认识提供参考。KD 的特征在于各种运动功能的缓慢进行性恶化,易被临床医师漏诊误诊,从而导致患者的生存质量下降且负担增加。临床医生应根据患者的临床表现和辅助检查作出鉴别诊断,早期诊断,并应尽快提供早期治疗。
伦理学声明:本研究方案经由河南科技大学第一附属医院伦理委员会审批(批号:2023-03-K0074),患者均签署知情同意书。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:柏亚真负责撰写论文、绘制图表;张登科负责文献、数据收集;杜敢琴负责论文设计、修改;富奇志负责指导撰写论文并最后定稿。
猜你喜欢
历史教学问题(2022年6期)2022-02-28
现代装饰(2021年5期)2021-12-02
中西医结合心脑血管病杂志(2016年20期)2016-03-01
汽车维护与修理(2016年3期)2016-02-28
现代电生理学杂志(2015年1期)2015-07-18
能源(2015年8期)2015-05-26
西安交通大学学报(医学版)(2015年2期)2015-02-28
汽车维护与修理(2015年6期)2015-02-28
汽车维护与修理(2015年5期)2015-02-28
环球时报(2012-02-07)2012-02-07
