
新发胚系TP53 突变致Li-Fraumeni 综合征相关急性B 淋巴细胞白血病1 例
2024-04-24 02:03:42徐启璐冉学红孙艳花刘军卞倩玉刘丽萍
中国肿瘤临床 2024年2期
徐启璐 冉学红 孙艳花 刘军 卞倩玉 刘丽萍
患者男性,13 岁。因持续性腰背部疼痛伴有间断性牙龈出血及鼻出血半个月于2020 年10 月就诊于潍坊市人民医院。血常规:白细胞(WBC)3.99×109/L,血红蛋白(HGB)101 g/L,血小板(PLT)55×109/L,C反应蛋白(CRP)138.3 mg/L。实验室检查:腹部彩超示肝左叶前后径8.0 cm,肝右叶斜径16.1 cm;脾大,20.4 cm×5.8 cm。骨髓形态:原始幼稚淋巴细胞占61%,外周血原始幼稚淋巴细胞占17%。免疫分型:异常B 淋巴细胞占有核细胞的14.7%,普通型急性B淋巴细胞白血病(acute B lymphoblastic leukemia,BALL)。骨髓染色体:46,XY[7]。骨髓病理:成熟骨小梁间见增生的异性淋巴样细胞,结合免疫组织化学符合B-ALL。基因检测:43 种白血病融合基因筛查:WT1/WT1 内参基因:0.15%。急性淋巴细胞白血病ALL(acute lymphoblastic leukemia,ALL)基因突变筛查(16 种):TP53 突变,一级变异,突变位点TP53:NM_000546:exon5:c.G524A:p.R175H,rs28934578,频率52.94%。患者完全缓解后多次复查第二代测序(NGS)均示TP53(p.R175H)突变阳性,变异等位基因频率(variant allele frequency,VAF)值持续稳定约在50%。对患者头发、指甲及口腔黏膜行TP53 基因突变检测,提取标本中基因组DNA,使用聚合酶链式反应(polymerase chain reaction,PCR)引物对p53 基因的全部编码序列及外显子与内含子拼接区进行扩增。将阳性样本与SAP 进行DNA 测序,测序后使用Chromas 测序分析软件进行分析。检测到TP53 基因Exon5 存在错义突变:c.524G>A(p.R175H),结合多个基因检测结果,考虑患儿TP53 突变为胚系突变。确证亲缘关系,患者一级亲属均不存在肿瘤病史,二级亲属中祖父58 岁因肺癌去世,外祖母50 岁诊断为乳腺癌。在征得监护人知情同意后,采集先证者父母、弟弟的外周血,根据标准DNA 提取方案提取DNA。使用PCR 和Sanger 测序法行TP53 基因Exon5:c.524G>A(p.R175H)突变位点检测;结果证明其父母、弟弟外周血分子分析显示纯合野生型等位基因。患儿的分子检测揭示了一种新发的胚系突变,该胚系突变导致TP53 的5 号外显子524 位点鸟嘌呤被腺嘌呤替代,导致175 位的精氨酸替换成组氨酸。此突变在他的父母、弟弟中均未发现。先证者所携带的TP53(p.R175H)突变来自其父或母的配子突变,见图1~3。本例患者符合李-佛美尼综合征(Li-Fraumeni syndrome,LFS)诊断标准,见图4。给予利妥昔单抗+环磷酰胺+长春地辛+多柔比星+地塞米松/甲氨蝶呤+阿糖胞苷(R+HyperCVAD/MA A+B 方案)诱导治疗1个周期,多参数流式细胞术(flow cytometry,FCM)测微小残留病灶(minimal residual disease,MRD)未见异常B 淋巴细胞,疗效评估达完全缓解(complete response,CR)。后再次行R+HyperCVAD/MA A+B 方案化疗3 个周期,长春地辛+柔红霉素+培门冬酶+泼尼松(VDLP 方案)化疗2 个周期,长春地辛+伊达比星+培门冬酶+地塞米松(VILP 方案)化疗3 个周期,依托泊苷+长春地辛+阿糖胞苷+地塞米松(EOAP 方案)化疗2 个周期,环磷酰胺+长春地辛+泼尼松+6-巯嘌呤(COMP 方案)化疗1 个周期;患者持续缓解1 年10 个月余,期间行腰穿鞘注治疗6 次,脑脊液检查正常。2022 年8 月患者疾病复发后给予VILP 方案诱导化疗2 个周期未缓解。采集自体淋巴细胞,予以地西他滨+阿糖胞苷化疗。患者自体细胞培养失败,给予氟达拉滨+环磷酰胺(FC 方案)清淋预处理,行CD19 的嵌合抗原受体NK 细胞(chimeric antigen receptor NK-cell,CAR-NK)治疗。共回输CD19 CARNK 细胞2 次,疾病未缓解。患者因新冠病毒感染暂无法行嵌合抗原受体T 细胞(chimeric antigen receptor T-cell,CAR-T)治疗,予奥加伊妥珠单抗(抗体偶联药物ADC 靶向CD22 抗原的单克隆抗体与一种细胞毒制剂卡奇霉素偶联而成)静脉输注2 次。治疗效果不理想。再次采集自体淋巴细胞,予地西他滨、FC 预处理,回输CD19 CAR-T 细胞共1 次,疾病未缓解。回输当天患者新型冠状病毒核酸检测阳性并高热,给予抗感染、托珠单抗治疗,后患者新冠肺炎重症感染,给予呼吸机辅助通气、奈玛特韦/利托那韦抗病毒、激素抗炎、联合抗感染、强心、利尿等支持治疗,患者出现细胞因子反应,呼吸、循环衰竭死亡。

图1 先证者TP53 基因 Exon5 c.524G>A(p.R175H)突变型测序图谱
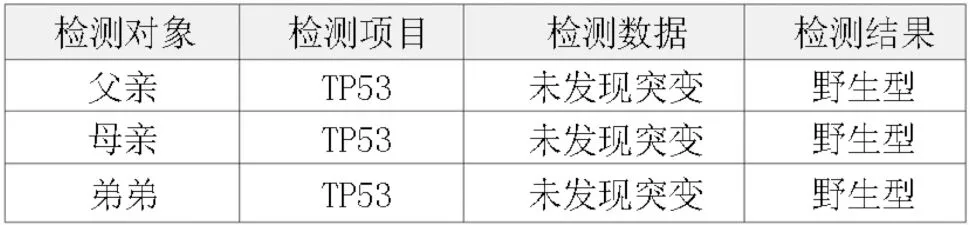
图2 TP53 基因野生型测序图谱

图3 遗传病家系中先证者及其父母基因突变与疾病关系的研究

图4 先证者家系图谱
小结胚系突变又叫生殖细胞系突变,是指发生在从受精卵到配子的生殖系中的突变。发生在与体细胞分化点之后的原始生殖细胞及其后代的突变,会传递给下一代,但在父母体细胞中不存在,通常被称为新发胚系突变[1-2]。新发胚系突变的发生可能导致个体遗传疾病的发生或后代罹患肿瘤的风险增加。约75%的TP53 胚系突变存在于具有遗传性癌症易感性的 LFS 家族中,是一种少见的常染色体显性遗传病[3],与27 个家族性癌症病史以及致病/可能致病的TP53胚系变异(P/LP TP53 28GV)有关。其特征在于多种肿瘤的早期发作[4]。美国国立综合癌症网络(NCCN)指南[5]中收录了两种诊断标准:经典的LFS 综合征标准需满足以下全部条件:先证者在45 岁之前确诊为肉瘤;一级亲属在45 岁之前确诊任何癌症;一级或二级亲属在45 岁之前确诊任何癌症或在任何年龄确诊为肉瘤。Chompret 标准进一步扩大其疾病范围并提出更新的诊断标准,具备以下任意一项条件即可:1)先证者46 岁前明确诊断为LFS 综合征谱系疾病(包括绝经前乳腺癌、骨与软组织肉瘤、中枢神经系统肿瘤、肾上腺皮质肿瘤、白血病或细支气管肺泡癌)中的任意一种,并至少有1 名一级或二级亲属56 岁前明确诊断为LFS 综合征谱系疾病(若先证者罹患乳腺癌则家系中不包括乳腺癌);2)先证者罹患多种肿瘤(除外多种乳腺肿瘤),并且其中2 种是LFS 综合征谱系疾病,最早发现的肿瘤于46 岁前发病;3)若先证者罹患肾上腺皮质肿瘤或脉络丛肿瘤,则家系中可患病或不患病。本例患者符合LFS 综合征的Chompret 诊断标准。LFS 综合征与较高的癌症发病率相关,儿童期至30 岁的相对发病率最高。累积终生癌症发病率接近100%。约50%的LFS 综合征患者在30 岁前会患上癌症,男性的终生风险高达75%,女性的风险几乎为100%,且乳腺癌的风险占比增加[1]。50%LFS 综合征患者在30 岁以前发生至少一种LFS 综合征相关癌症,而一般人群中30 岁以前的癌症发病率为1%,并且易于患治疗诱导的继发性癌症[2]。LFS 综合征中白血病的发病率为0~4%,包括亚二倍体的ALL 和治疗相关的髓系肿瘤。亚二倍体的ALL 与TP53 突变相关,与低总生存率和高复发率有关,预后非常差[3]。TP53胚系突变患者移植效果差,本例先证者其一级亲属均不适合行造血干细胞移植(hematopoietic stem cell transplantation,HSCT),因此有必要选择突变基因相关靶向治疗,Chen 等[6]首次报道了使用CAR-T 细胞疗法治疗LFS 综合征背景下的复发/难治性B-ALL患者,患者接受了抗CD19 和抗CD22嵌合抗原受体CAR-T 细胞“鸡尾酒”疗法,并通过形态学和FCM 估达到CR,MRD 阴性。该患者复发后再次接受了新一轮抗CD22 CAR-T 细胞治疗后仍然有效。因此,对于传统化疗和HSCT 无效的LFS 综合征相关的复发难治性血液系统恶性肿瘤,CAR-T 细胞疗法可能是一种很好的替代选择。本例患者因新冠病毒感染未能显现出CAR-T 疗效。
综上所述,TP53 胚系突变是儿童遗传性肿瘤易感的重要突变基因,不断鉴定出新发胚系突变的事实强调了TP53 基因突变分析的重要性,家系中检测出TP53 胚系突变的未患病者,可行定期体格、血液及影像学检查,利于早期发现肿瘤,同时针对TP53 基因的靶向治疗能够帮助临床医生更好管理这种遗传性肿瘤患者,改善预后。
本文无影响其科学性与可信度的经济利益冲突。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
家庭医学(下半月)(2020年1期)2020-05-11 02:05:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
海峡姐妹(2018年7期)2018-07-27 02:30:36
特别健康(2018年4期)2018-07-03 00:38:08
特别健康(2018年2期)2018-06-29 06:13:42
重庆医学(2015年12期)2015-03-05 05:52:54
