
HPLC-MS/MS法测定调味粉中5种罂粟碱含量的不确定度评估
2024-04-13 11:04于晓慧石梦璇程凤丽王晓
食品工业 2024年3期
于晓慧 ,石梦璇,程凤丽,王晓
1.山东特检科技有限公司(济南 250000);2.济南富美特信息科技有限公司(济南 250000)
含有吗啡、可待因等20多种生物碱的罂粟壳,国家市场监管总局已明确禁止在食品中使用[1]。长期食用,会对人体产生很大的伤害,但是,一些商家为达到盈利目的,仍不择手段,在一些调味粉和调味料中加入,能够增加食物的美味,赢得消费者的青睐[2]。当前检测吗啡、可待因等生物碱含量的方法主要有高效液相法[3-5]、气相色谱质谱法[6-7]、液相色谱质谱联用法[8-11]、薄层色谱法[12]、气相色谱法。用液相色谱质谱联用方法测定吗啡、可待因、罂粟碱、那可丁和蒂巴因,具有灵敏度高,操作简单,上机溶液不需要进行衍生操作等优点。试验选用高效液相色谱-串联质谱法测定调味粉中5种罂粟壳生物碱。
不确定度的评定是保证试验分析过程准确性和可靠性的重要手段[13-14],试验依据CNAS-GL006—2019《化学分析中不确定度的评估指南》,建立高效液相色谱-串联质谱法测定调味粉中的吗啡、可待因、罂粟碱、那可丁和蒂巴因含量的数学模型,分析在含量测定的整个过程中引入不确定度的分量和占比,得到对试验数据结果产生较大影响的因素,从而为在实际应用场景中使用此方法的操作过程提供合理建议,同时提高调味粉中的吗啡、可待因等5种罂粟壳生物碱测定结果的准确性和可靠性,为评价检验报告提供科学依据。
1 材料与方法
1.1 仪器与试剂
LC-MS/MS 8045三重四极杆液质联用仪(岛津公司);JA31002型电子天平(上海舜宇恒平科学仪器有限公司);BK-180J型超声波清洗器(山东博科仪器有限公司);UMV-1型多管旋涡混合器(北京优晟联合科技有限公司);KL04A型离心机(湖南凯达科学仪器有限公司)。
乙腈(色谱纯,Thermo Fisher);实验室用水(一级水,屈臣氏);甲酸(色谱纯,Thermo Fisher);调味粉(超市购买);5种罂粟壳生物碱标准品、可待因标准品(北京坛墨质检科技有限公司)。
1.2 混合标准品溶液的制备
1.2.1 标准品储备液
精密移取1 mL吗啡、可待因、罂粟碱、那可丁和蒂巴因混标对照品(其中吗啡、可待因质量浓度25 μg/mL,罂粟碱、那可丁和蒂巴因浓度5 μg/mL)于同一10 mL容量瓶中,加乙腈溶解并定容至刻度,摇匀,作为混合标准品储备液,于4 ℃冰箱避光保存,备用。
1.2.2 同位素内标工作液(5 μg/mL)
分别精密吸取吗啡-D3同位素溶液和可待因-D3同位素对照品(50 μg/mL)各1.0 mL,置于10 mL容量瓶中,用乙腈定容至刻度,配制成吗啡-D3、可待因-D3浓度均为5.0 μg/mL的混合内标工作溶液。于4 ℃冰箱中避光保存备用。
1.2.3 系列标准工作溶液
精密移取20,40,80,160,400和800 μL标准储备液于10 mL容量瓶中,分别精密加入同位素内标工作溶液(5.0 μg/mL)40 μL,用乙腈定容至刻度,得到吗啡、可待因质量浓度为5,10,20,50,100和200 ng/mL,罂粟碱、那可丁和蒂巴因质量浓度为1,2,4,10,20和40 ng/mL系列标准溶液,内标质量浓度20 ng/mL,现用现配。
1.3 供试液的制备
称取2 g(精确至0.01 g)超市购买的某品牌调味粉于50 mL聚四氟乙烯具塞离心管中,加入1~3 mL水,涡旋振荡30 s,用移液管准确加入15 mL乙腈,密塞,涡旋1 min,超声处理30 min,取出,加入6 g无水硫酸镁和1.5 g无水醋酸钠的混合粉末,立即涡旋振荡2 min,使吸附样品中的全部水分,以4 000 r/min离心5 min,取上清液,以0.22 μm滤膜过滤,取滤液适量,进样,待测定。
1.4 检测方法
1.4.1 色谱参考条件
色谱柱Shim-pack GIST C1-AQ HP(100 mm×2.1 mm,1.9 μm);仪器柱温40 ℃;流动相流速0.30 mL/min;仪器进样量2 μL;流动相A为0.1%甲酸水,B为乙腈。梯度洗脱条件:0.5~1.5 min,10%~70% B;1.5~3.5 min,70%~95% B;3.5~5.5 min,95% B;5.5~5.6 min,95%~10% B;5.6~8.0 min,10% B。
1.4.2 质谱条件
离子源为电喷雾离子源;扫描方式为正离子扫描;采集方式MRM;离子源接口电压+4.5 kV,-3.5 kV;碰撞气为氩气;离子传输管温度250 ℃;接口温度300 ℃;加热模块温度400 ℃。表1为5种罂粟碱物质的质谱参数。

表1 化合物的质谱参数
2 结果与分析
2.1 不确定度分析
2.1.1 数学模型调味粉中5种化合物含量计算模型如式(1)所示。
式中:X为调味粉样品中吗啡、可待因、罂粟碱、那可丁和蒂巴因含量,μg/kg;c为标准曲线上查得的样品质量浓度,ng/mL,V为定容体积,mL;m为取样量,g;
2.1.2 识别和分析不确定度的来源
如图1所示,HPLC-MS/MS法测定调味粉中吗啡、可待因、罂粟碱、那可丁和蒂巴因含量引入的不确定度主要包括称量调味粉样品引入的不确定度、标准溶液配制引入的不确定度、调味粉样品前处理引入的不确定度、样品重复性试验、加标回收试验、计算过程中的标准曲线拟合带来的不确定度和SHIMADZU LC-MS/MS 8045三重四极杆液质联用仪引入的不确定度。
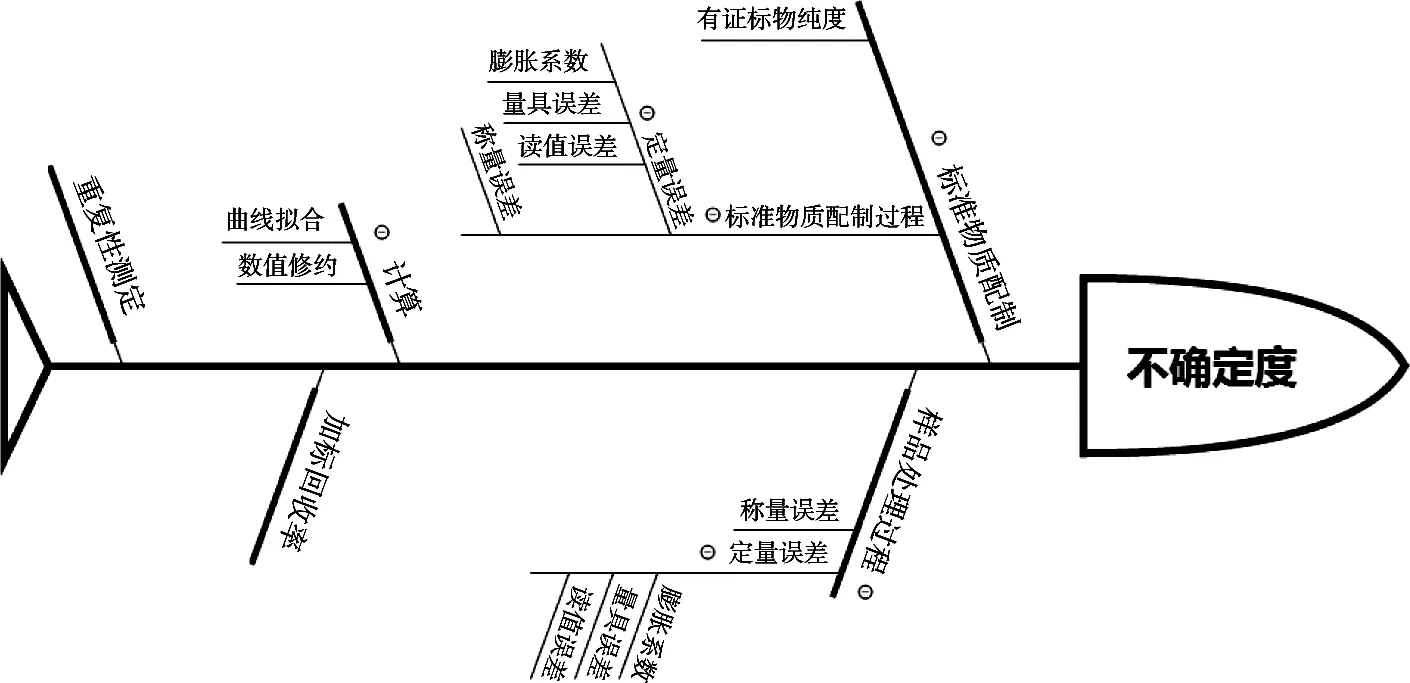
图1 不确定度的来源分析图
2.2 分析不确定度分量评定
2.2.1 标准品引入的相对标准不确定度urel(s)
2.2.1.1 外标标准品纯度引入相对标准不确定度urel(1)
由吗啡和可待因的标准品证书得,两者扩展不确定度为0.05 μg/mL,k=2;罂粟碱、那可丁和蒂巴因的标准品证书可得两者扩展不确定度为0.01 μg/mL,5种标准品纯度引入的相对标准不确定度分别为urel(11)=0.05/(25×2)=0.001,urel(11)=0.01/(5×2)=0.001。
2.2.1.2 内标标准品纯度引入相对标准不确定度urel(2)
根据吗啡-D3同位素溶液和可待因-D3同位素对照品标准物质证书得,扩展不确定度为3 μg/mL,k=2,则urel(21)=3/(50×2)=0.030,由内标标准品引入的相对标准不确定度为
2.2.1.3 标准品储备液配制引入相对标准不确定度urel(3)
在5种化合物标准品溶液配制过程中所用玻璃器具引入的不确定度包含2个。一是温度引起不确定度,配制吗啡和可待因标准溶液时,环境温度为25 ℃,JJG 196—2006《常用玻璃量器检定规程》检定A级玻璃容器的温度为20 ℃,所以这里考虑的是液体体积膨胀,配制过程中使用乙腈定容,乙腈的膨胀系数为0.001 37,按照均匀分布进行计算,温度引起液体体积变化产生的不确定度为其中V为玻璃器具的规格。二是玻璃器具区间半宽a产生的不确定度,按照均匀分布产生的不确定度标准不确定度urel(V)=u(V)/V,相对标准不确定度u(V)=按照上述评定方法,在配制标准品储配液过程使用到的A级玻璃容器有10 mL容量瓶2次和1 mL单线移液管2次,标准储备液过程中引入的相对标准不确定度urel(3)结果为0.025 49,具体计算结果见表2。
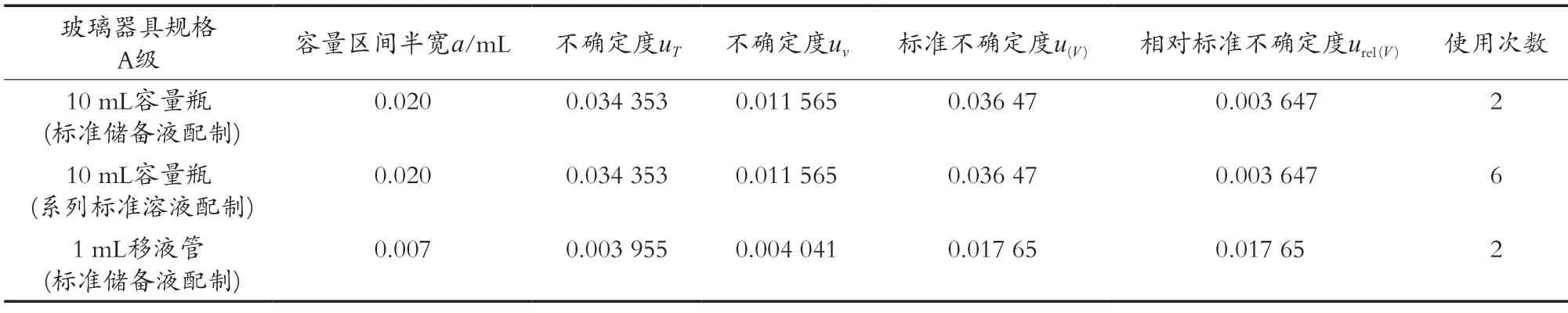
表2 器具引入的不确定度
2.2.1.4 系列标准溶液中引入相对标准不确定度urel(4)
在系列标准溶液配制的过程中用到20~200 μL移液器、100~1 000 μL移液器和10 mL容量瓶。以20~200 μL移液器为例,根据JJG646—2006《移液器检定规程》规定,使用20~200 μL移液器,移取液体体积20 μL时,最大容量允差为±20×2.0%,按照矩形分布,取K=则20~200 μL移液器(移取体积20 μL)容量允差产生的标准不确定度为0.230 9 μL;已知乙腈的膨胀系数为0.001 37,实验室温度为20±5 ℃,由温度产生的标准不确定度为0.079 1 μL,则由20~200 μL移液器(20 μL)产生的相对标准不确定度为0.011 55。
配制该系列标准工作溶液过程中使用6次10 mL容量瓶(A级)、20~200 μL移液器10次和100~1 000 μL移液器2次,经计算系列标准溶液配制过程中引入的相对标准不确定度urel(4)结果为0.040 63,计算结果见表3。

表3 移液器不同体积引入的不确定度
2.2.1.5 标准物质的相对标准不确定度urel(s)
表2和表3显示配制标准储备液和系列标准工作液时,所用到的玻璃器和移液器所引入的不确定度。标准品引入的相对标准不确定度公式为计算得到5种化合物由标准品引入的相对标准不确定度结果,见表4。
表3 移液器不同体积引入的不确定度

表4 标准品引入的不确定度
2.2.2 样品前处理过程引入的相对标准不确定度urel(y)
前处理过程中带来的不确定度主要是由调味粉样品的称量引起的,称量样品使用的天平规格是万分之一,该天平的允许误差是0.5 mg,按照均匀分布计算,样品前处理过程引入的相对标准不确定度为
2.2.3 样品重复性试验引入的相对标准不确定度urel(f)
称取6份同一供空白试品,分别加入64 μL的标准品储备液、60 μL同位素内标工作液(5 μg/mL),在1.3的前处理条件下进行提取供试液,在1.4的检测条件下进行测定。根据公式计算,重复性标准偏差为重复性标准不确定度为重复性试验引入相对标准不确定度为计算结果见表5。

表5 重复性试验引入的不确定度
2.2.4 回收率试验引入过相对标准不确定度urel(r)
称取同一本底空白调味粉试样6份,分别加入64 μL的罂粟碱混合标准品储备液、60 μL同位素内标工作液(5 μg/mL),在1.3的前处理条件下进行提取供试液,在1.4的检测条件下进行测定计算,得回收率标准偏差Sr,回收率引入的标准不确定度为相对标准不确定度为结果见表6。

表6 回收率试验引入的不确定度
2.2.5 拟合曲线引入相对标准不确定度urel(l)
标准曲线采用最小二乘法拟合,测定系列标准曲线级别为6,测定样品次数6次,计算残差标准偏差用最小二乘法拟合标准工作曲线求得样品中含量过程所引入的标准不确定度公式为相对标准不确定度计算得5种罂粟壳类生物碱在标线拟合过程中相对标准不确定度,见表7。

表7 标准曲线合成引入的不确定度
标准不确定度公式中:SA为标准溶液残差标准偏差;n为标点的个数;p为测定样品的次数;c样为测量样品的浓度;为系列标准溶液平均浓度;b为标准曲线斜率。
2.2.6 仪器引入的相对标准不确定度urel(i)
由岛津LC-MS/MS 8045三重四极杆液质联用仪器的校准证书可知,urel(i)为2%,按照均匀分布计算,k=2,得仪器引入的相对标准不确定为urel(i)=2%/2=0.01。
2.3 不确定度的合成和扩展
2.3.1 不确定度的合成
根据得到的各个不确定度分量,对相对标准不确定度进行合成,公式为经计算得到吗啡等五种生物碱的相对标准品不确定度分别为0.058 31,0.063 68,0.053 49,0.071 83和0.079 24。
2.3.2 不确定度的扩展
取置信区间为95%(k=2),U=urel(x)×2x,计算得到调味粉中5种待测物质的扩展不确定度结果分别为8.96,9.72,1.69,2.27和2.49 μg/kg。
2.3.3 结果表示
通过高效液相色谱-串联质谱法测定调味粉中吗啡、可待因、罂粟碱、那可丁和蒂巴因,测定结果分别为76.87±8.96,76.35±9.72,15.77±1.69,15.78±2.27和15.71±2.49 μg/kg。
3 结论
通过对HPLC-MS/MS法测定调味粉中吗啡、可待因、罂粟碱、那可丁和蒂巴因这5种罂粟碱化合物的含量测定进行不确定度评估。结果显示,在整个检测过程中,比较各个分量的相对标准不确定度,影响比较大的是标准溶液的配制和标准曲线的拟合。在实际的工作中,要求检测人员优化移取和定容的具体细节,并且定期对试验人员进行试验操作和基础理论培训,提高检测结果的稳定性和准确度。通过不确定度的研究,可为调味粉中罂粟碱的检测提供参考和借鉴。
猜你喜欢
中国动物保健(2022年2期)2022-05-05
实用手外科杂志(2021年4期)2022-01-05
技术与市场(2020年5期)2020-03-02
中国药物滥用防治杂志(2020年4期)2020-01-09
实验与分析(2018年3期)2019-01-04
时代英语·高一(2018年5期)2018-11-19
实验与分析(2018年4期)2018-02-27
时代英语·高一(2017年5期)2017-11-14
实用手外科杂志(2015年4期)2015-08-27
中国药业(2014年21期)2014-05-26
