
Cryptic divergences and repeated hybridizations within the endangered “living fossil” dove tree (Davidia involucrata) revealed by whole genome resequencing
2024-04-12 02:04YumengRenLushuiZhngXuchenYngHoLinYupengSngLndiFengJinqunLiuMinghuiKng
植物多样性 2024年2期
Yumeng Ren ,Lushui Zhng ,Xuchen Yng ,c,Ho Lin ,Yupeng Sng ,Lndi Feng ,Jinqun Liu ,b,**,Minghui Kng ,b,*
a Key Laboratory of Bio-resource and Eco-environment of Ministry of Education,College of Life Sciences,Sichuan University,Chengdu 610065,China
b State Key Laboratory of Herbage Improvement and Grassland Agro-ecosystem,College of Ecology,Lanzhou University,Lanzhou 730000,China
c Guangdong Provincial Key Laboratory of Plant Adaptation and Molecular Design,Guangzhou Key Laboratory of Crop Gene Editing,Innovative Center of Molecular Genetics and Evolution,School of Life Sciences,Guangzhou University,Guangzhou 510006,China
Keywords: Davidia involucrata Cryptic lineage Hybridization Population genomics Positive evolution
ABSTRACT The identification and understanding of cryptic intraspecific evolutionary units (lineages) are crucial for planning effective conservation strategies aimed at preserving genetic diversity in endangered species.However,the factors driving the evolution and maintenance of these intraspecific lineages in most endangered species remain poorly understood.In this study,we conducted resequencing of 77 individuals from 22 natural populations of Davidia involucrata,a “living fossil” dove tree endemic to central and southwest China.Our analysis revealed the presence of three distinct local lineages within this endangered species,which emerged approximately 3.09 and 0.32 million years ago.These divergence events align well with the geographic and climatic oscillations that occurred across the distributional range.Additionally,we observed frequent hybridization events between the three lineages,resulting in the formation of hybrid populations in their adjacent as well as disjunct regions.These hybridizations likely arose from climate-driven population expansion and/or long-distance gene flow.Furthermore,we identified numerous environment-correlated gene variants across the total and many other genes that exhibited signals of positive evolution during the maintenance of two major local lineages.Our findings shed light on the highly dynamic evolution underlying the remarkably similar phenotype of this endangered species.Importantly,these results not only provide guidance for the development of conservation plans but also enhance our understanding of evolutionary past for this and other endangered species with similar histories.
1.Introduction
The question of how biodiversity evolves is a fundamental inquiry in the fields of evolutionary and conservation biology(Schluter,2000;Seehausen,2009).A crucial prerequisite for understanding the evolution of biodiversity in natural populations is the accurate definition of evolutionarily distinct and conservation units (Crandall et al.,2000).This is essential for preserving biological diversity,as it provides managers and policymakers with a clear understanding of the boundaries of population units for endangered species (Funk et al.,2012).Understanding the genetic diversity of endangered species holds significant importance in conservation biology.Despite their morphologically similar appearance,cryptic lineages may have evolved within a single endangered species,and each lineage deserves conservation efforts due to their distinct adaptations to different environments(Bolnick et al.,2003;Bickford et al.,2007;Palsbøll et al.,2007).Neglecting such lineages can introduce bias into predictions regarding their responses to environmental change and biodiversity loss(Scheffers et al.,2012;Pauls et al.,2013;Adams et al.,2014;Feckler et al.,2014).Therefore,accurately determining conservation units can prevent both the underestimation of the necessary protection units for endangered species and the misallocation of resources on abundant species(Frankham et al.,2010).The recognition of cryptic lineages has become increasingly apparent due to advancements in molecular techniques (Moritz,1994;Petit et al.,1998;Pfenninger and Schwenk,2007;Wang et al.,2009;J¨orger and Schr¨odl,2013;Shang et al.,2015;Torres-Cambas et al.,2017;Feng et al.,2019).Beyond their implications for conservation,the presence of such lineages and hybridizations also challenges our understanding of population connectivity(Pante et al.,2015),speciation(Seehausen,2009)and even ecosystem functioning(Brodersen and Seehausen,2014).
The deep intraspecific lineages were found to have emerged as a result of both geographical and climatic changes in various species(Wang et al.,2009,2015;Li et al.,2021;Hu et al.,2022;Sang et al.,2022;Shen et al.,2022;Wu et al.,2023).For instance,the Quaternary glaciation and the accompanying cooling climates drove intraspecific divergences in different refugia for plants and animals alike (Hewitt,2000;Qiu et al.,2011).However,postglacial range expansions further resulted in the meeting of the diverged lineages and subsequent hybridizations (Zachos et al.,2001;Petit et al.,2003;Hewitt,2004).Additionally,with each deep intraspecific lineage,long-term selection may favor the most suitable genotypes that adapt to local habitats,which can be detected through population genomic data (Zhang et al.,2020;Hu et al.,2022).For the ancient plants known as “fossil” relics,which originated early and left behind ancient fossils,it is likely that past geographic and climatic fluctuations played a significant role in driving both interspecific and intraspecific divergences (Mao and Liu,2012).A notable example can be found within the fossil genusCercidiphyllum,where two lineages,representing two distinct species,diverged approximately 5 million years ago (Mya) during the Pliocene epoch.This period coincided with significant geographic and climatic changes in eastern Asia(Qi et al.,2012).Moreover,it is probable that subsequent demographic fluctuations and hybridization events occurred between these lineages in response to the later climatic changes (Zhu et al.,2020).These events likely led to the introgression of both chloroplast and nuclear DNA (Qi et al.,2012).Apart from the genusCercidiphyllum,many more fossil genera are found in this region,such asGinkgo(Zhao et al.,2019),Euptelea(Cao et al.,2020),Tetracentron(Liu et al.,2020),andDavidia(Tang et al.,2017).Understanding how past geographic and climatic fluctuations have influenced the divergence and hybridization of these fossil genera over their extended presence in this region is of great interest.These evolutionary events have the potential to complicate conservation efforts and challenge our understanding of the formation of hidden lineages (Allendorf et al.,2010;Naciri and Linder,2015).Additionally,it is crucial to identify the key genes that may have played a significant role in the local adaptation of these distinct lineages in the genomic era (Hu et al.,2022).In general,regions of the genome that have experienced positive or adaptive selection exhibit significantly greater genetic differentiation compared to other regions that have evolved neutrally.Therefore,we can identify selection signals by examining the “outliers” of theFSTvalues,which serve as an index of genetic differentiation between populations (Zhao et al.,2019;Zhu et al.,2020).Gene-environment association analyses address the issue of environmental heterogeneity across different populations and aim to establish relationships between patterns of allele frequencies and environmental gradients (Sang et al.,2022;Shen et al.,2022).By employing these two approaches,we can gain a better understanding of the process and potential of adaptation.
The only species of the genusDavidia,Davidia involucrata,is found exclusively in southwestern and central China in eastern Asia(He et al.,2004;Yang et al.,2019).It thrives in humid,rainy,and foggy evergreen and deciduous mixed forests but is sensitive to drought stress(Zhang et al.,2000;Liu et al.,2019).Detailed reports on the geographic distribution and eco-physiological characteristics of this species suggest that it has developed local adaptations(Tang et al.,2017;Yang et al.,2020).Its unique white bracts,which resemble doves,have earned it the name “dove tree” and contribute to its high ornamental value (He et al.,2004).This species is considered as a Cenozoic relict plant endemic to China(Tang et al.,2017)and is often referred to as the“giant panda”of the plant world.While it was once widely distributed in North America and East Asia,the Quaternary glaciation event caused a significant decline in its population size(Eyde,1997;Manchester et al.,2009).The relic rarity and limited distribution of natural populations have led to the classification ofD.involucrataas a nationally protected first-class wild plant in China (Liu et al.,2019).The fruits ofD.involucrataare dispersed by animals,which limits their longdistance dispersal (Zhang et al.,2000).The seeds of this species experience a dormancy period of 2-3 years,with a very low germination rate (Song and Bao,2006).D.involucratais an entomophilous species,likely pollinated by bees and other insects(Sun et al.,2008).Extensive studies have reported high genetic diversity and inter-population differentiation in this endangered species using molecular methods and genetic analysis methods (Song and Bao,2006;Luo et al.,2011;Li et al.,2012;Chen et al.,2015;Ma et al.,2015).Two major cryptic lineages have been identified: one in central China and the other in southwest China (Chen et al.,2015;Ma et al.,2015).However,one population in southwest China had a chloroplast haplotype closely related to the eastern lineage (Chen et al.,2015).These two lineages were estimated to have diverged 4-5 Mya (Chen et al.,2015;Ma et al.,2015).Nuclear SSR markers revealed multiple inter-lineage introgressions due to long-distance gene flow (Ma et al.,2015).
In this study,we conduct whole-genome resequencing on 77 individuals ofDavidia involucrata,including 67 newly sequenced individuals,from its known natural distribution range in China.Our objective is to examine cryptic divergence and hybridization within this relic “fossil” plant.We identify one more cryptic lineage in addition to two major lineages previously identified.We also investigate whether certain genes exhibited positive signals during local adaptation and assessed the inbreeding levels and mutation load of these lineages and populations.Through these analyses,our aim is to evaluate evolutionary units,providing valuable insights for the conservation and management of this species.
2.Materials and methods
2.1.Plant sampling and genome resequencing
In addition to 10 previously sequenced genomes (Chen et al.,2020),we added whole-genome resequencing data of 67 individuals representing 22 natural populations within the known distribution range of dove-tree in central and southwest China.For each population,individuals were sampled at least 100 m apart from each other.Silica gel-dried leaves of 67 individuals were collected for DNA extraction in this study (Table S1).All of the samples were sequenced using DNBSEQ-T7 platforms with a sequencing depth of 20× .
2.2.Data filtering and single nucleotide polymorphism (SNP)calling
Raw reads were filtered using Fastp v.0.20.0(Chen et al.,2018)to remove adapters and low quality base.Reads with a “N” base number of 5 bp and reads containing more than 40% of mass value ≤20 bases were removed: cropping with a 4 bp sliding window with an average mass below 20.The quality-filtered reads were mapped to theDavidia involucratareference genome (Chen et al.,2020) with BWA-MEM v.0.7.12 (Li and Durbin,2009) and sorted with Samtools v.1.10 (Li et al.,2009).BaseRecalibrator and ApplyBQSR in GATK v.4.1.1.7 (McKenna et al.,2010) were used to calculate the distribution of systematic error and correct base quality according to the base quality value of BAM file.Finally,single nucleotide polymorphism(SNP)calling was performed using HaplotypeCaller,CombineGVCFs and GenotypeGVCFs of GATK to generate a VCF file with the parameter “all sites” (including nonvariant sites).To reduce the false positive error,SNPs were further filtered with VCFtools v.0.1.13 (Danecek et al.,2011) as following steps: (1) Sites with a quality score lower than 30 and SNP sites within 5 bp near InDel were filtered out.(2) Remove SNPs with more than two alleles.(3)Sites with extremely high(less than onethird of the average depth)coverage and extremely low(more than threefold of the average depth) coverage were treated as missing sites.(4)Filter out sites with more than 20%percentage missing and minimum allele frequency (MAF)<0.05.
2.3.Population structure and phylogenetic analysis
Phylogenetic and population structure analyses require the use of SNPs with low selection pressure and a true response to natural variation in order to obtain an accurate topology of the phylogenetic tree.We used VCFtools to convert the data to a binary file as an input file for PLINK v.1.07 (Purcell et al.,2007),linked loci was removed using the parameters: -indep-pairwise 50 5 0.2.We constructed a maximum likelihood (ML) phylogenetic tree with FastTree v.2(Price et al.,2010)software in the use of 1,722,440 LDpruned,evolution-information SNPs.We searched for single copy of lineal homologous gene ofCamptotheca acuminata(Kang et al.,2021),Nyssa yunnanensis(Mu et al.,2020),Nyssa sinensis(Yang et al.,2019) andDavidia involucrata(Chen et al.,2020),with protein sequences,then OrthoFinder2 (Emms and Kelly,2019) software was used to classify the gene families of the protein sequence,distinguish the orthologous and paralogous genes,and screen out the orthologous gene families with only one copy in each of the four species,which was regarded as the single-copy orthologous genes ofD.involucratafor subsequent analysis.Python script was used to extract all SNPs within the Coding sequence(CDS)interval of these single copy orthologous genes from the SNP-merged VCF file based on the extracted single copy gene information.The GTR-GAMMA model of RAxML v.8.2.11 (Alexandros and Stamatakis,2014) was used to establish the single copy of orthologous genes extracted from 4 species,and the bootstrap support was repeatedly estimated using 100 bootstraps.It is then visualized using iTOL (https://itol.embl.de,Letunic and Bork,2007).
We used 1,722,440 independent SNPs among 77 samples for Principal component analysis(PCA) using PLINK v.2018 and population structure inferred using ADMIXTURE v.1.3.0 (Alexander and Lange,2011) with K-value setting ranging from 1 to 5.For further insight into relationships among lineages,we performed identityby-descent blocks analysis after missing genotype estimation using the algorithm from BEAGLE v.4.1 (Browning and Browning,2007)with the following parameters: window=100,000;overlap=10,000;ibdtrim=150;ibdlod=15.
2.4.Genetic diversity
We eliminated the ancestry-mixed individuals inferred from the analysis of population structure.Nucleotide diversity (π),quantified population genetic differentiation (FST),and Tajima's D between each pair of groups were calculated using VCFtools v.0.1.17.All of these three values were calculated using 20 kb nonoverlapping sliding window.The π was estimated after taking into account both polymorphic and monomorphic sites.We measured and compared patterns of linkage disequilibrium(LD)for each group using PopLDdecay v.3.4.1 (Zhang et al.,2019).
2.5.Demographic history inference and gene flow
We selected“pure”individuals in structure analysis(Q>0.9999)with sequencing coverage (>15×) to estimate the demographic history of effective population size (Ne) over time using the pairwise sequential Markovian coalescent (PSMC) method (Li and Durbin,2011).For each lineage,we selected 2 individuals for PSMC analyses with 100 bootstrap replicates.The mutation rate was set as 1.87 × 10-9per base per year,and generation time was set as 20 years following Chen et al.(2020).At the same time,VCFtools v.0.1.13 software was used to eliminate deviation from haven-equilibrium(0.001)SNPs for downstream analyses.We used a perl script(vcf2maf.pl;https://github.com/wk8910/bio_tools/)to generate two-dimensional joint SFS (2D-SFS) with “pure” individuals.Further,we used FASTSIMCOAL2(Excoffier et al.,2021)to infer the differentiation time and gene flow among the lineages ofDavidia involucrata.To reduce the influence of natural selection,we only used SNPs in intergenic region for historical population dynamics simulations.We then designed and simulated 8 different evolutionary models based on their genetic structure(Fig.S5) and 10 different evolutionary models for different gene flow scenarios(Fig.S6) in FASTSIMCOAL2.For each model,we performed 50 independent runs with 100,000 coalescence simulations per likelihood estimation and 40 cycles of the likelihood maximization algorithm to search the global ML parameter estimates.The best model was identified through the Akaike's information criterion(AIC).Identical to PSMC,we set a mutation rate of 3.74× 10-8per base per generation and a generation time of 20 years.
2.6.Identification of environment-associated SNPs
Two methods were used to identify genotype-environment association (GEA) loci across the whole-genome.We kept common SNPs with MAF >10%,including a total of 10,608,122 for analyses.Firstly,a univariate latent-factor linear mixed model(LFMM)in the R package LEA v.3.3.2 (Frichot and François,2015) were implemented to search for associations between allele frequencies and the 19 BIOCLIM environmental variables (Fick and Hijmans,2017).Three latent factors to account for population structure in the genotype data based on the number of ancestry clusters inferred with ADMIXTURE v.1.3.0 in LFMM analyses.For each environmental variable,five independent Markov chain Monte Carlo(MCMC)runs were conducted by using 5000 iterations as burn-in followed by 10,000 iterations.We used false discovery rate FDR correction of 5%for the significance cutoff.Secondly,we performed a multivariant redundancy analysis (RDA) to search for candidate loci with a low false-positive rate.We selected four uncorrelated environment variables(BIO2,BIO4,BIO17,BIO18)with a correlation<0.7 before running RDA analyses using the R packageveganv.2.6-2(Oksanen et al.,2017).The overlapped genetic loci by both approaches were regarded as “core adaptive loci” for local adaptation.
For functional annotation,Davidia involucratapredicted proteincoding genes were aligned to multiple public databases including Swiss-Prot,NR,using NCBI BLAST+v.2.2.31 with an E-value of 1e-5as the cutoff.The highest bit score was used to select the best homologous genes for comparison,so as to obtain the specific function of genes.InterProScan software was used to compare structural databases such as InterPro database and Pfam to predict the existing domains in the genome(Zdobnov and Apweiler,2001).In addition,we then enriched the GO function of the core adaptive sites with R packagetopGO(Alexa and Rahnenfuhrer,2022).
We used neutral (the 1,722,440 LD-pruned SNPs used for population structure analyses) and adaptive(the 22,630 core adaptive loci)variants to calculate correlation between genetic distance(FST/(1-FST))and geography(IBD;Mantel test) and environmental (IBE;partial Mantel test,after excluding geography influence),respectively.Geography and environmental distance(Euclidean distance)accounted by latitude and longitude of the samplings with significance determined using 999 permutations in the R packagevegan.
2.7.Genes with positive selection signals
To identify genomic regions potentially under selection inDavidia involucrata,we calculated values of π ratio,FST,XP-CLR(Chen et al.,2010) between West lineages and East lineages(comprising genetic “pure” individuals in ADMIXTURE analyses)using a genome-wide sliding windows strategy (20 kb sliding windows and 0 kb steps) in VCFtools v.0.1.13.After calculating all tests,the windows with more than 100 SNPs andPvalues less than 0.025(right side-Z test)were considered as significant outliers.The windows meet three conditions simultaneously were considered under significant selective pressure.To further investigate the annotation of core adaptive variants,we used the core adaptive variants identified inD.involucratato run blastp v.2.10.0+(Camacho et al.,2009) against the protein libraries ofArabidopsis thaliana.The best alignment for each gene was kept and considered to be homologous.Then,we searched for annotation of the homologs inA.thalianain The Arabidopsis Information Resource(TAIR) database (https://www.arabidopsis.org) to obtain the annotations of the core adaptive variants.
2.8.Mutation load analyses
Whole genome heterozygosity (He) is the proportion of heterozygous sites in the whole genome that exclude gaps.For each sample,He was computed based on an in-house python script.The run of homozygosity (ROHs) was calculated using PLINK.We calculated the proportion of the genome (0-1) that is in runs of homozygosity (ROHs) (FROH) for each individual.To test whether these isolated populations experience inbreeding depression,we estimated the relative excess of derived loss-of-function (LoF) and missense variants in 22 populations.We employed methods previously used for gorillas(Xue et al.,2015)to measure the mutation load.SnpEFF v.4.3 (Cingolani et al.,2012) was used to create the gene function database based on the gene annotation file of theD.involucratagenome.Then,the high-quality SNPs were classified into different functional classes (synonymous,missense,loss of function)by SnpEFF with default settings.Finally,we calculated the total number of mutations of each class by counting each heterozygous genotype once and each homozygous alternative genotype twice.
We inferred the ancestral and derived allele at each location based on comparison to the genome of their close relativesCamptotheca acuminataandNyssa sinensis.The probability of the derived versus ancestral allelic state were inferred using est-sfs v.2.03(Keightley and Jackson,2018).The ancestral allele was set as the non-deleterious allele,and the derived allele as the potential deleterious allele.We calculated the ratio of missense and loss-offunction to synonymous mutations at the homozygous and heterozygous sites.
3.Results
3.1.Population structure and genetic diversity
A total of 1.2 Tb of whole genome sequencing data were generated from 77 samples of 22 populations,with an average sequencing depth of 19.57× (Fig.1A and Table S1).Using the chromosome-levelDavidia involucratareference genome (Chen et al.,2020),the average mapping rate of the raw reads was 97.12%,and the average genome coverage rate was 93.78%(Table S1).After filtering,16,363,317 SNPs (MAF >0.05) were retained for subsequent analyses.

Fig.1.Population genomic analyses of Davidia involucrata.(A) Sample locations of sampled D.involucrata populations.Charts at sampling locations indicate the distribution of genetic groups identified by ADMIXTURE analysis when K=3.The map was retrieved from Google Earth(https://www.google.com/earth/).Elevation data for the map were derived from SRTM elevation data through the WorldClim data website(https://www.worldclim.org/).(B)Population structure bar plots.The scenarios of K=2-4 was shown,and K=3 is the best value according to cross-validation analysis.(C) Principal component analysis (PCA) plots showing the first two principal components.
Population genetic structure analysis by ADMIXTURE revealed that when K=2,all individuals were divided into two distinct clusters.The western cluster comprised individuals from Gansu,Sichuan,Yunnan,and western Guizhou of southwest China,while the eastern cluster included individuals from eastern Guizhou,Hubei,Hunan,Shaanxi of central China.The individuals from the southern region of southwest China showed ancestral admixture(Fig.1B).Approximately,89.39% of the genetic composition of this third lineage was shared with the western lineage,while the remaining 10.61% was shared with the eastern lineage.Crossvalidation error analysis suggested that the optimal classification was three genetic groups (K=3) (Table S2).When K=3,individuals of this southern lineage from western Guizhou were further separated from the remaining populations of the western lineage (Fig.1C).We identified a wide hybrid zone (Hybrids1) between the western and southern lineages,as well as another hybrid zone(Hybrids2)between the southern and eastern lineage,both of which were located in the Sichuan Basin.Principal component analysis(PCA)exhibited a similar differentiation pattern among the three groups (Fig.1C).The first principal component (PC1;explained variance=11.53%) separated the western lineage from the eastern lineage,while the second principal component (PC2;explained variance=5.46%)further divided them into five groups:the western lineage,the Hybrids1 group,the southern lineage,the Hybrids2 group,and the eastern lineage.
To construct phylogenetic tree,the maximum likelihood method was employed using 1,722,440 SNPs,with aNyssa sinensisindividual serving as an outgroup(Fig.S1A).A total of 5267 singlecopy orthologous genes were identified based on the protein sequences ofN.yunnanensis,N.sinensis,Camptotheca acuminataandDavidia involucrata.Similarly,maximum likelihood method was used to construct phylogenetic trees for single-copy orthologous genes (Fig.S1B).The phylogenetic trees constructed by SNPs and single-copy genes yielded consistent results(Fig.S1).
To assess patterns of genetic differentiation and genetic diversity across the genome,genetically admixed individuals were excluded,and parameters such as genetic diversity(π),LD patterns,and Tajima's D were calculated.The π values of each lineage ranged from 2.23×10-3to 2.84×10-3,with the eastern lineage exhibiting the highest diversity and the southern lineage showing the lowest diversity (Fig.S2).Pairwise genome-wide averages genetic divergence (FST) values among the three groups ranged from 0.255 to 0.422 (Table S4).TheFSTvalues between the eastern lineage and western or southern lineage were notably higher than that between the western and southern lineages (Table S4),indicating consistent genetic cluster patterns observed in ADMIXTURE,PCA,and NJ tree analyses.When assessing pairwiseFSTamong populations,a high level of differentiation was observed amongD.involucratagroups: the western lineage VS the eastern lineage(meanFST=0.421),the southern lineage VS the eastern lineage(meanFST=0.422).However,the mean pairwiseFSTbetween the western lineage and the southern lineage was significantly lower(0.255).The mean pairwise nucleotide differences in inter-lineage comparisons (dxy) ranged from 0.0112 to 0.0137 (Table S4),showing a same pattern.Tajima's D values varied from 0.5414 to 1.1786 (Fig.S3).Positive Tajima's D value may indicate a common historical demographic bottleneck inD.involucrata.The level of genetic diversity and LD decay revealed different demographic histories among the three lineages.The southern lineage exhibited more link imbalance,a slower LD decay rate (Fig.2A),lower nucleotide diversity,and may have experienced a long-term population bottleneck or recent demographic expansions resulting in a decreased genetic diversity.
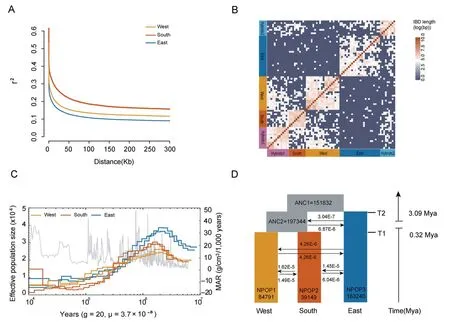
Fig.2.Demographic history of Davidia involucrata.(A) Patterns of linkage disequilibrium for the three lineages.(B) Estimated haplotype sharing between individuals.Heatmap colors represent the total length of identity-by-descent blocks for each pairwise comparison.(C)Inferred demographic history of the West(yellow lines),South(orange lines),and East (blue lines) populations inferred by the pairwise sequentially Markovian coalescent (PSMC) method over the past 10 million years.(D) The best fitting model (model10)diagram and simulated parameters with maximum likelihood values are obtained using FASTSIMCOAL2 software,and estimated effective population size and differentiation time are given.The numbers next to the arrows represent mobility per generation between populations.
3.2.Demographic histories and gene flow
We used the pairwise sequentially Markovian coalescent(PSMC;Li and Durbin,2011)to reconstruct the demographic history of each lineage.All three lineages exhibited similar demographic histories,characterized by a high effective population size (~1.2 Mya) followed by a bottleneck around 0.8 Mya (Fig.2C).From the Pliocene to the early Pleistocene,all lineages experienced population expansion,reaching their maximum effective population sizes(Fig.2C).The eastern lineage underwent three expansions and two contractions.The first expansion occurred in the late Miocene and early Pliocene,with the effective population size peaking at 38,000 around 2 Mya,followed by a decline (the first contraction),which coincided with the increase in the mass accumulation rate(MAR)of loess in China.Subsequently,the population expanded from 0.07 Mya to 22 thousand years ago (Kya) (the second expansion) and experienced a contraction during 10-20 Kya (the second contraction).After the population expanded again around 10 Kya(the third expansion),the effective population size remained stable at 10,000.The western lineage experienced two expansions and one contraction,while the southern lineage experienced two expansions and two contractions.From 10 Mya to 1.1 Mya,the effective population size of the western lineages increased (the first expansion),reaching a maximum around 1.1 Mya (25,000 and 30,000,respectively),and then continuously declined (the first contraction).The effective population size of the western lineage declined continuously(the first contraction)from 1.1 Mya to 20 Kya,remained stable between 0.1 Mya and 0.3 Mya,increased (second expansion) thereafter,and remained constant at around 7000 during 20-10 Kya.The southern lineage reached its maximum effective population size of 30,000 around 1.05 Mya,followed by a decline from 1.05 Mya to 30 Kya(the first contraction),and then an increase during 30-10 Kya (the second expansion).
We employed a coalescent simulation-based method using fastsimcoal2 to estimate the timing of divergence and demographic histories of the three groups.We constructed ten models to represent the divergence of the three groups,and the best-fit model was determined based on the lowest AIC value and highest likelihood (Tables S7 and S8).The best-supported model indicated that the eastern lineage diverged from the common ancestor of the western and southern lineages during the Late Pliocene,approximately 3.09 Mya,while the divergence between the western and southern lineages occurred during the Middle Pleistocene,around 0.32 Mya (Fig.2D).The simulations also revealed different rates of gene flow among the three lineages.Low levels of ancient gene flow were estimated,showing significant asymmetric gene flow between the common ancestor of the western and southern lineages and the eastern lineage(MANC2←E=3.04 × 10-7,ME←ANC2=6.87 × 10-6),with greater gene flow from the ANC2 to the eastern lineage compared to the reverse direction.The primary direction of gene flow was from the common ancestor (ANC2) of the western and southern lineages to the eastern lineage.Furthermore,after divergence of the western and southern lineages,estimates of gene flow between the two lineages were low and symmetrical (MS←W=1.49 × 10-5,MW←S=1.62×10-5),and the same was observed for western and eastern lineages (ME←W=4.26 × 10-6,MW←E=4.26 × 10-6).The gene flow between the latter two lineages was lower(ME←S=6.04 × 10-6,MS←E=1.45 × 10-5),with a greater gene flow from the eastern to the southern lineage compared to the reverse direction.
3.3.Identification of gene variants associated with local adaptation
We employed two genotype-environment association (GEA)methods,LFMM and RDA,to identify gene variants associated with environmental factors across the total distribution range ofDavidia involucrata.Using LFMM,we tested the GEA of 19 environmental variables (Table S9) and identified 311,096 SNPs significantly associated with one or more environmental variables,based on a qvalue cutoff of 0.05(Fig.3A).The RDA method,which detects GEA SNPs associated with multivariate environments,was performed using four uncorrelated variables (BIO2,BIO4,BIO17,and BIO18)selected to avoid issues related to multicollinearity(Fig.S7).A total of 439,424 SNPs were identified by RDA,with 22,630 SNPs overlapping with LFMM (Fig.3A).These shared SNPs were considered“core adaptive variants” for local adaptation and were associated with 2881 genes (gene region and upstream/downstream 2 Kb)(Fig.3A).
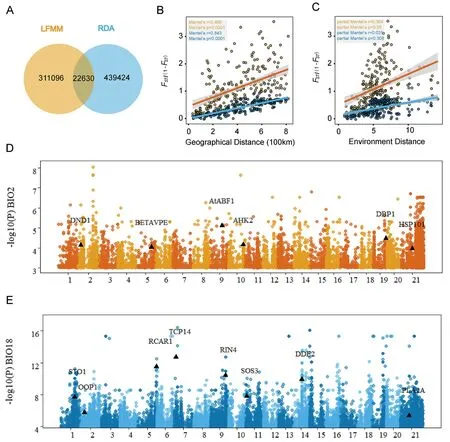
Fig.3.Genome-wide screening of the loci associated with local environmental adaptation.(A)Genotype-environment association(GEA)genetic loci explored by latent factor linear mixed model(LFMM)and redundancy analysis(RDA).(B)Isolation-by-distance analyses(Mantel test,two-sided)for populations(n=77)based on neutral(dark blue dots and light blue line) and adaptive variants (yellow dots and orange line) separately.The shadow of linear regression denotes the 95% confidence interval.(C) Isolation-by-environment analyses (partial Mantel test,two-sided,controlling for the effect of geographic distance) for populations (n=77) based on neutral (dark blue dots and light blue line) and adaptive variants(yellow dots and orange line)separately.The shadow of linear regression denotes the 95%confidence interval.(D,E)Manhattan plot for variants associated with the Mean Diurnal Range (BIO2) (red) and the Precipitation of the Warmest Quarter (BIO18) (blue).Selected candidate genes are labeled in the plot at their respective genomic positions.
To assess the patterns of isolation by distance(IBD)and isolation by environment (IBE)in neutral and potentially adaptive SNPs,we conducted Mantel and partial Mantel tests (Fig.3B and C).The results showed a significant pattern of IBD in both neutral and adaptive variants (Fig.3B).However,the partial Mantel test revealed a significant IBE only in adaptive SNPs,indicating that the genetic variation of adaptive variants was primarily influenced by the environment (Fig.3C).Furthermore,we performed gene ontology (GO) enrichment analysis of the core adaptive genes,which revealed enrichment in growth and metabolic pathways,suggesting their role in environmental adaptation inDavidia involucrata(Fig.S8 and Table S10).
To provide examples of significant and well-knownArabidopsis thalianahomologous genes identified in associated with representative climate variables,we selected BIO2 (temperature) and BIO18 (precipitation) (Fig.3D and E and Table S11).For example,HSP101(Dinv16319),associated with BIO2,is a cytosolic heat shock protein required for acclimation to high temperature and essential for plant survival under heat stress (Babbar et al.,2023).AHK2(Dinv21174) regulate plant organ size,flowering time and plant longevity(Bartrina et al.,2017).Additionally,there are other genes involved in immunity (e.g.DND1andCAD1),pollen development and fertility (CER22andDRP1C),embryogenesis (EMB2775),ABA responses (AtABF1andCYP707A4).STO1(Dinv13307),associated with BIO18,encodes 9-cis-epoxycarotenoid dioxygenase,encodes a key enzyme in the biosynthesis of abscisic acid.It catalyzes the first step of ABA biosynthesis from carotenoids and in doing so,enabling plant response to water stress (Al-Younis et al.,2021).Other precipitation-associated genes,such asPUB23(Dinv43996) andATHB29(Dinv16647),are also involved in the response to water stress.Additionally,there are genes functionally involved in abscisic acid signaling regulation (e.g.ABI3andRCAR1),immunity(e.g.RIN4andWRKY18),and growth and development (e.g.IAA22andPUR4).
3.4.Positively selected genes within the eastern or western lineage
Due to the low population and individual numbers in the southern lineage,we focused our analyses on identifying positively selected genes in the eastern and western lineages.We analyzed the population genomic data of the “pure” individuals,excluding the hybrid populations,using metrics such as the π ratio,interspecific differentiation(FST),and XP-CLR.In the western lineage,we discovered a total of 28 protein-coding genes homologous toArabidopsis thaliana,which were located in 15 outlier windows (20 kb) exhibiting selection signatures (Fig.4A and Table S12).Among these genes,several are associated with stress response,reproduction,development,and metal transport(Figs.4C and S9).For instance,HSP60-2(Dinv12114)is involved in copper ion and ATP binding and plays a role in the inflammatory response and salt stress inA.thaliana(Velinov et al.,2020).Another gene,MEE70(Dinv19349),encodes a WD-40 repeatcontaining protein that participates in chromatin assembly as part of the CAF1 and FIE complex.Mutants ofMEE70exhibit parthenogenetic development,including the proliferation of unfertilized endosperm and embryos (Chen et al.,2023).PGM2(Dinv28052)encodes a cytosolic phosphoglucomutase (PGM),and the loss of bothPGM2andPGM3significantly impairs male and female gametophyte development (Malinova et al.,2014).VAN(Dinv37124) encodes a homeodomain transcription factor with sequence similarity to theArabidopsisovule development regulator geneBell1.Mutants ofVANexhibit additional lateral organs and phyllotaxy defects (Bencivenga et al.,2016).Lastly,ATAMT1(Dinv11772) encodes a plasma membrane-localized ammonium transporter and contains a cytosolic trans-activation domain essential for ammonium uptake (Fig.S9).
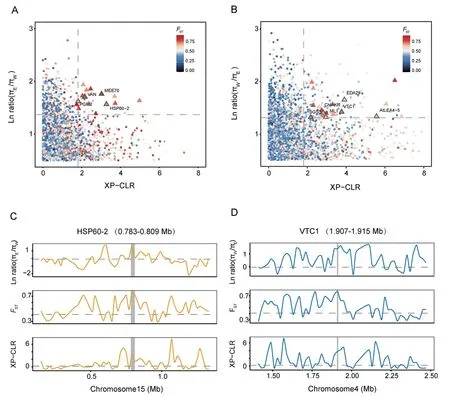
Fig.4.Genomic region with selection signals.(A)Distribution of the XP-CLR(x-axis),ln ratio πE/πW(y-axis)and value of pairwise fixation index(FST)(color)between West and East lineages.The dashed vertical and horizontal lines indicate the significance threshold(corresponding to Z-test,P<0.025,where XP-CLR>1.8122,ln ratio πE/πW>1.369859,and FST>0.6785626)used for extracting outliers(triangle symbol).(B)Distribution of the XP-CLR(x axis),ln ratio πW/πE(y axis)and value of pairwise fixation indices(FST)(color)between West and East lineages.The dashed vertical and horizontal lines indicate the significance threshold (corresponding to Z-test, P <0.005,where XP-CLR >1.8122,ln ratio πW/πE>1.299672,and FST>0.6785626)used for extracting outliers(triangle symbol).(C)Selective sweep on chromosome 15(0.783-0.809 Mb).(D)Selective sweep on chromosome 4(1.907-1.915 Mb).Horizontal dashed lines represent the whole genome mean for the corresponding parameters.
In the eastern lineage,we identified a total of 36 protein-coding genes located in 20 outlier windows that have signals of positive selection (Fig.4B and Table S12).Several of these genes are associated with stress resistance,disease resistance,reproduction,and development(Figs.4D and S10).One of the genes identified isVCT1(Dinv35561),which is involved in cell wall carbohydrate biosynthesis,protein glycosylation,and ascorbate (vitamin C) biosynthesis.VCT1may play a role in controlling the transcription of defense-related and senescence-associated genes and influence ammonium sensitivity (Zhang et al.,2022).SGS3(Dinv07279) is required for posttranscriptional gene silencing and natural virus resistance (Mourrain et al.,2000).ACS(Dinv07280) appears to function as a monomer and may have an important role in preventing the toxic accumulation of fermentation products,including acetaldehyde,acetate,and ethanol (Mou et al.,2020).CHlAKR(Dinv35557) is believed to play a role in detoxifying reactive carbonyl compounds that could impair the photosynthetic process(Treffon et al.,2022).AtLEA4-5(Dinv14293) is involved in protecting enzyme activities from the adverse effects induced by freeze-thaw cycles in vitro(Cuevas-Velazquez et al.,2021).EDA29(Dinv31762) regulates phytochrome and responses to continuous far-red light stimulus through the high-irradiance response system(Staneloni et al.,2009).
3.5.Heterozygosity,inbreeding level and mutation load of Davidia involucrata
We conducted an analysis of whole-genome heterozygosity,inbreeding level,and mutation load in the three defined groups.The heterozygosity values ranged from 2.20 × 10-3to 5.87 × 10-3(Fig.5A).The eastern lineage exhibited the highest heterozygosity(meanHe=4.63×10-3)and the largest population size,while the southern lineage showed the lowest heterozygosity (meanHe=3.18 × 10-3) and the smallest population size.The western lineage displayed a moderate level of heterozygosity (meanHe=3.63 × 10-3) and population size.

Fig.5.Diversity,linkage disequilibrium,and mutation load metrics for Davidia involucrata.(A)Box plot of heterozygosity of the whole genome.(B)Box plot of FROH(sum of ROH>10 kb/genome effective length)for each individual in the two species.The line in the center of the box represents the median values,the edges of the box represent the first and third quartiles,and the whiskers above and below the box show the range of values.(C) The ratio of loss-of-function (LoF) variations in homozygous and heterozygous related to synonymous variations.(D) The ratio of missense variations in homozygous and heterozygous related to synonymous variations.
The decrease in heterozygosity observed in allDavidia involucrataindividuals may be associated with high inbreeding coefficients.Genomic inbreeding coefficients (F) ranged from 0.12 to 0.33 (Fig.5B).Individuals with a higher degree of inbreeding generally had lower heterozygosity and higher levels of continuous pure sum fragments.The southern lineage exhibited the highest level of genomic inbreeding (mean F=0.2354),followed by the western lineage (mean F=0.1873) and the eastern lineage (mean F=0.1594) (Fig.5B).These findings indicate that the southern lineage had a higher overall inbreeding level compared to the western lineage,and the western lineage had a higher inbreeding level compared to the eastern lineage.
To assess the mutation load of each lineage,we calculated the ratio of deleterious derived mutations to synonymous derived mutations (Del/Syn) and the ratio of loss-of-function derived mutations to synonymous derived mutations (LoF/Syn).The homozygous Missense/Syn and LoF/Syn values of derived alleles were highest in the South lineage (Fig.5C and D),suggesting that the southern lineage accumulated a greater mutation load compared to the eastern and western lineages.We also calculated the Missense/Syn and LoF/Syn values for derived alleles in both homozygous and heterozygous genotypes across different lineages.The Missense/Syn and LoF/Syn values were higher in the heterozygous form compared to the homozygous state.This may be due to the fact that homozygous deleterious mutations are more likely to cause individual death,resulting in higher fitness for heterozygous mutations compared to homozygous mutations.Consequently,most harmful mutations are present in the population in heterozygous genotypes.The proportion of alleles derived from homozygous genotypes and alleles derived from heterozygous genotypes was highest in the southern lineage (LoF/Syn).The eastern lineage exhibited the highest proportion of alleles derived from heterozygous genotypes,while the proportion of alleles derived from homozygous genotypes in the eastern lineage was similar to the western lineage and lower than that of the southern lineage.The western lineage had a low proportion of alleles derived from both homozygous and heterozygous genotypes compared to the other lineages.This pattern of mutation load suggests that the southern lineage experienced a more prolonged population bottleneck and thus carried more alleles derived from homozygous genotypes,indicating a higher risk of sustained decline in effective population size.
4.Discussion
In this study,we identified three cryptic local lineages within the endangered “fossil” plantDavidia involucratabased on wholegenome data from representative populations across its entire distribution region.Additionally,we observed hybrid populations that resulted from gene flow between the three lineages.We discovered numerous gene variants correlated with the environment and also identified many genes showing signals of positive evolution in the local lineages.These findings indicate the highly dynamic evolution occurring within this species despite its similar phenotype.Our results are crucial for the development of new conservation strategies for this endangered species.
4.1.Cryptic divergence and hybridization during the long evolutionary history
The current phenotype ofDavidia involucrataclosely resembles its Cenozoic fossil counterpart(Eyde,1997;Manchester et al.,2009;Tang et al.,2017).This lineage is estimated to have diverged from the monotypic genusCamptothecaapproximately 60 Mya (Chen et al.,2020).If no other species underwent further evolution and differentiation for the genusDavidia,D.involucratahas survived since its split fromCamptothecafor a considerable period.It was once distributed in North America,with populations or distinct lineages there extinguished (Eyde,1997;Manchester et al.,2009).The relic populations in central and southwest China may now represent only a fraction of the once widely distributed populations and lineages.Our analysis,based on whole genomic data,indicates that the earliest divergence between the eastern lineage and the common ancestor of the western and southern lineages occurred around 3 Mya(Fig.2).This falls within the Pliocene epoch,although it is younger than the estimates of 4-5 Mya from two earlier studies that relied on several DNA markers only(Chen et al.,2015;Ma et al.,2015).Furthermore,our analysis suggests that the divergence between the western and southern lineages took place around 0.3 Mya(Fig.2).These findings indicate that all of the other lineages or populations originated before the Pliocene may have become extinct,as suggested by fossil records (Eyde,1997;Manchester et al.,2009).The current three lineages originated from further divergence whenD.involucrataretreated to central and southwest China.The Pliocene to Quaternary period witnessed significant geographical and climatic fluctuations (An et al.,2001),which likely contributed to the formation of the three observed lineages.Interestingly,intraspecific divergences have been discovered during this period in other “fossil” plants,such asCercidiphyllum japonicum(Zhu et al.,2020),Euptelea pleiospermum(Cao et al.,2020),andTetracentron sinense(Liu et al.,2020).Therefore,it is highly probable that many plants in this region experienced similar geographical and climatic influences that facilitated their intraspecific local divergences.
We further discovered that these divergences between lineages involved the selection of allelic variations in many genes related to environmental adaptation.For instance,we identified allelic variations in 2881 genes that were correlated with environmental factors(Fig.3A).These genes are enriched in growth and metabolic pathways (Fig.S8 and Table S10).In the eastern lineage,we observed signals of positive selection in many genes associated with stress resistance,disease resistance,reproduction,and development (Figs.4B-D and S10;Table S12).In the western lineage,we identified several genes with selection signatures(Figs.4A-C and S9;Table S12) that are linked to stress response,reproduction,development,and metal transport (Fig.3C).These genetic variations likely provided the basis for the local adaptation and the following divergence ofDavidia involucrata.Despite these localized selections,we still observed multiple hybridization events between the three lineages.Between the eastern and southern lineages,we identified two distinct hybrid populations in the areas between their respective distributions.Additionally,two populations at the northern distribution range of the western lineage,with a significant geographic gap,exhibited clear introgressions from the eastern lineage (Fig.1).Four populations located in the intermediate distribution range between the western and southern lineages clearly originated from hybridization between them.This hybrid zone extended over a relatively wide range.Our demographic analyses of the three lineages (Fig.2) suggested that gene flow between them has persisted from their divergence to the present.These frequent hybridizations contrast with previous studies (Chen et al.,2015;Ma et al.,2015) that were based on a limited number of DNA fragments and only detected minimal gene flow between the eastern and western lineages.Moreover,this finding is inconsistent with the limited gene flow inferred from both seed dispersal(Zhang et al.,2000) and pollinators(Sun et al.,2008).The existence of unknown seed and pollination dispersal animals responsible for long-distance dispersals should be further investigated.
It is important to note that the third southern lineage demonstrated genetic admixture from both the western and eastern lineages when K=2 (Fig.1).All sampled individuals of two populations showed the stable genomic compositions from the other two lineages,indicating that they may have evolved as one independently evolving lineage for many generations after the initial hybridization as modelled by our coalescent analyses(Fig.2).This differs from those partly introgressions only a few individuals and unstable genetic admixtures between individuals in the hybrid populations.However,our coalescent analyses of the alternative origin models still support its bifurcating divergence from the western lineage,with substantial introgression from the eastern lineage.This is likely due to approximately 89% of genomic elements originating from the western lineage.On the other hand,the hybrid lineages with a composition of 75%from one parent and 25%from the other parent unequivocally indicate their origin from hybridization rather than a bifurcating divergence (Wang et al.,2021).This situation warrants further investigation,particularly regarding the extent to which a small genomic contribution from the lineage influenced the establishment and maintenance of the southern lineage through reproductive isolation from hybridization(Wang et al.,2021).One caveat in our study,only two populations were sampled this southern lineage.More populations should be added in the future analyses.
4.2.Mutation load and conservation implication
Among the three recovered lineages,the southern lineage exhibited the lowest diversity,possibly due to the small number of individuals sampled and their restricted distribution range.We discovered evidence of inbreeding in each lineage,as indicated by the presence of continuous homozygous segments ranging from 0.12 to 0.33(Fig.5B).This inbreeding can result in the accumulation of deleterious mutations,known as mutation load,which is primarily influenced by heterozygous recessive deleterious mutations.The southern lineage demonstrated a relatively higher mutation load,while the eastern lineage displayed a comparatively lower level of mutation load.The accumulation of mutation load is closely associated with the population dynamics history,including factors such as effective population size,population expansion and contraction,and the duration of population bottlenecks (Robinson et al.,2023).The southern lineage may have experienced rapid population expansion or bottlenecks,with the effective population size remaining around 1000 individuals(Fig.2A).This would result in the loss of rare alleles,and subsequent genetic drift and inbreeding events would increase the rate of fixation of homozygotes,converting heterozygous recessive deleterious mutations into homozygotes(Lynch et al.,1995).On the other hand,both the eastern and western lineages experienced a significant decrease in effective population size,but they later rebounded and maintained relatively large effective population sizes (Fig.2).However,it is important to note that the genetic diversity and all examined mutation load are significantly higher or lower than those observed in relic trees based on genomic data from eastern Asia(Chen et al.,2019;Cao et al.,2020;Liu et al.,2020;Zhu et al.,2020).
The species level diversity θπ values and θWvalues ofDavidia involucratawere obtained based on resequencing data(θπ=4.67×10-3;θW=1.16×10-3;Table S3).Through comparison,it was found that the genetic diversity level ofD.involucratawas higher than that of other “fossil” plants:Ginkgo biloba(θπ=2.11 × 10-3,θw=2.36 × 10-3) (Zhao et al.,2019);Chinese lineage ofCercidiphyllum japonicum(θπ=1.05 × 10-3) (Zhu et al.,2020);Liriodendron chinenseCW lineage (θπ=6.89 × 10-4),CE lineage (θπ=5.39 × 10-4) (Chen et al.,2019).Our population genomic analyses ofD.involucrataindicate relatively high genetic diversity.This suggests that despite being an endangered species,it does not face significant extinction pressure.One of the main reasons for this may be its outcrossing breeding system and the observed long-distance gene flow.Furthermore,gene flow and hybridization may contribute to the fitness of this endangered relic species.However,in addition to the two previously identified cryptic evolutionary lineages (Chen et al.,2015,2020;Ma et al.,2015),we have identified a third lineage in southern Sichuan and northern Yunnan.All three lineages should be effectively conserved as they represent distinct units,and attention should also be given to the hybrid populations between them.These hybrid populations have the potential to evolve into new evolutionary units,similar to what has been observed for the southern lineage.Additionally,priority should be given toin situimprovements in population size of each lineage and each population.It is recommended to protect the species in its natural habitat to maintain a relatively large effective population size and gene flow among the different lineages and populations.The conservation of the southern lineage should be prioritized.Forex situconservation,germplasm resources from all three lineages and hybrid populations should be collected to preserve the maximum genetic diversity of this fossil plant.
Data availability
The whole-genome resequencing data generated in this study have been deposited in the National Genomics Data Center(NGDC,https://ngdc.cncb.ac.cn) under accession number PRJCA020962.
CRediT authorship contribution statement
Yumeng Ren:Writing -review &editing,Writing -original draft,Software,Formal analysis,Data curation,Conceptualization.Lushui Zhang:Investigation.Xuchen Yang:Formal analysis.Hao Lin:Formal analysis.Yupeng Sang:Formal analysis.Landi Feng:Formal analysis.Jianquan Liu:Writing -review &editing,Methodology,Funding acquisition.Minghui Kang:Writing -review &editing,Writing -original draft,Project administration.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the Second Tibetan Plateau Scientific Expedition and Research program (No.2019QZKK0502),Strategic Priority Research Program of Chinese Academy of Sciences(No.XDB31010300),Fundamental Research Funds for the Central Universities,and International Collaboration 111 Program(BP0719040).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pld.2024.02.004.

- 植物多样性的其它文章
- Global patterns and ecological drivers of taxonomic and phylogenetic endemism in angiosperm genera
- Patterns and drivers of plant sexual systems in the dry-hot valley region of southwestern China
- Enhanced and asymmetric signatures of hybridization at climatic margins: Evidence from closely related dioecious fig species
- Cryptic diversity and rampant hybridization in annual gentians on the Qinghai-Tibet Plateau revealed by population genomic analysis
- Circumscription of the East Asia clade(Apiaceae subfamily Apioideae)and the taxonomic placements of several problematic genera
- Historical biogeography and evolutionary diversification of Lilium(Liliaceae): New insights from plastome phylogenomics