
Leber遗传性视神经病变罕见表型
——Harding病1例
2024-04-09 05:15:44王江南李雪景樊朕宏
中华眼视光学与视觉科学杂志 2024年3期
王江南 李雪景 樊朕宏
作者单位:河北医科大学第二医院眼病中心,石家庄 050000
患者,青年男性,因“双眼视力突然下降1 个月,伴双下肢无力6个月”于2023年1月2日就诊于河北医科大学第二医院眼病中心。眼科检查示双眼视力均为0.08,矫正不能提高,双眼瞳孔对光反射迟钝,相对性传入性瞳孔反应缺陷(-);眼底检查可见视盘充血,颜色鲜红,余未见明显异常。神经系统检查见双下肢肌力4 级,无肢体感觉异常,未引出病理征。双眼视野检查显示:右眼视野中心暗点,左眼中心及颞下方视野缺损。双眼视觉诱发电位(1,0 deg)示右眼N75-P100:5.7 μV;P100-N135:4.8 μV;P100潜伏期中度延长:139.7 ms;左眼N75-P100:4.1 μV;P100-N135:3.6 μV;P100潜伏期轻度延长:133.9 ms。双眼眼底血管荧光造影未见明显异常荧光。眼眶MRI显示双眼视神经T2WI呈节段性高信号,左侧视神经显著(图1A),增强后视神经T1WI略强化(图1B-D)。患者虽平素喜好吸烟(烟龄5年,每天约20支)和饮酒(酒龄10年,每天白酒250 g),但维生素D、叶酸测定及免疫相关抗体等实验室检查均未见异常。诊断:视神经脊髓炎谱系疾病(Neuromyelitis optica spectrum disorders,NMOSD)、Leber遗传性视神经病变(Leber's hereditary optic neuropathy,LHON)可疑。建议患者完善中枢神经脱髓鞘相关抗体检查及基因检测。中枢神经脱髓鞘相关抗体检查结果显示:抗AQP4抗体IgG、抗MOG抗体IgG、抗胶质纤维酸性蛋白抗体IgG均为阴性,但基因检测结果仍需20 d才可回报,鉴于患者神经系统异常表现,神经内科会诊讨论后予以口服甲泼尼龙片60 mg抗炎治疗(患者体质量60 kg),治疗期间,患者视力无明显改善,但双下肢肌力逐渐恢复至正常。激素快速减量并于1个月后停用,停用后半个月患者出现双下肢行走困难,再次就诊,神经内科查体显示:双下肢肌力3级,四肢腱反射(+),左侧脚踝以下浅感觉减退,右侧深感觉异常。次日四肢腱反射(++),2 d后近一步加重:双膝关节以下浅感觉减退,左侧深感觉异常。眼部检查显示:双眼视力进一步下降至指数/30 cm,双眼视觉诱发电位提示各波潜伏期较前进一步延长,光学相干断层扫描显示双眼视网膜神经纤维层厚度颞侧变薄。再次检查头颈部MRI显示,双眼视神经与图1比较,无明显改变,而颅脑MRI平扫、颈髓胸髓MRI平扫、胸部CT均未见明显异常,甲状腺功能检测、淋巴细胞计数检测、细胞因子检测等免疫相关检测均未见异常,排除感染等激素应用禁忌,与患者沟通后再次予以激素治疗。
治疗期间,患者及其姐姐基因检测回报(图2),均发现11778G>A位点突变,患者姐姐目前未出现临床症状,且发现患者神经系统临床表现并不符合AQP4-IgG阴性的NMOSD的诊断标准[1],加之患者眼部表现及同时合并多发性硬化表现,诊断为Harding病,排除NMOSD。遂快速减量激素,并给予辅酶Q10、复合维生素及艾迪苯醌(900 mg/d,口服治疗),随访近10个月,患者眼部情况未进一步加重,下肢肌力及感觉部分恢复。
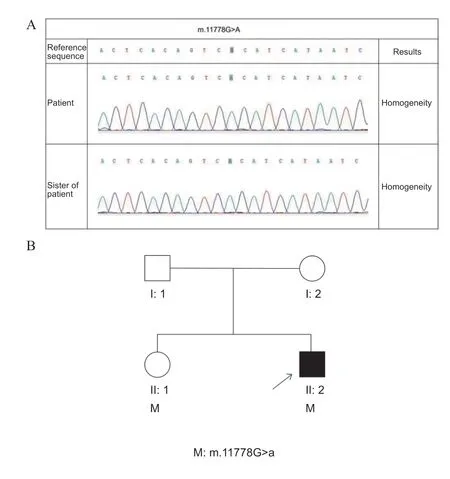
图2.Harding病患者及其姐姐线粒体基因测序结果和家系验证结果Figure 2.The results of mitochondrial gene sequencing and pedigree validation of the patient with Harding's disease and his sister
讨论
1871 年德国医生Theodor Leber 首次发现并报道LHON,超过90%的患者由 m.11778G>A、m.14484T>C、m.3460G>A这3 个原发突变引起[2]。1964 年临床上发现LHON与多发性硬化症(Multiple sclerosis,MS)共存的罕见关联。但直至20世纪90年代这种重叠表型才被Harding教授正式提出并命名为Harding病[3],该类患者以m.11778G>A位点突变为主[4],大多是女性(女男比为2.1∶1),发病年龄、视力丧失的严重程度和持续性与LHON相似,但多是单侧视力丧失,即使双眼受累,发病时间间隔也长于LHON,平均为20 个月[5]。Harding病神经系统损伤临床表现多样化,可以表现为轻微的神经系统障碍,部分可表现出持续性肌力减退[6],或典型的复发缓解性MS,甚至需要激素冲击治疗的侵袭性MS表现[7],像本研究报告中病例所示。目前认为是多种因素共同作用的结果,包括一个额外的X位点、线粒体呼吸链活性受损以及环境因素的影响,比如吸烟和饮酒也会诱发本病[8]。
Harding病的影像学可表现为典型的MS病变,二者相比,Harding病影像学中病变分布更明显,脑室周围病变沿白质束延伸,在侧脑室前角周围也有高信号[9],但也有一些病例报道提示存在非典型病变,大小和形状不同,边缘不清晰,T2序列亮度降低,T1序列[4]不可见。该患者眼眶MRI中有类似MS的脱髓鞘表现,但未在侧脑室等区域见到类似的高信号表现,推测可能与病灶较小有关。
在治疗方面,已有研究证明伊德贝酮有利于患者视力和视觉诱发电位的改善[10],米托蒽醌也与Harding病患者的视力恢复相关。虽然目前对于Harding病已有相关的治疗药物,但是由于该类患者合并的神经症状的多样性和复杂性,治疗方案仍然应根据个案的情况来决定。另有研究团队发现mtDNA突变和线粒体功能障碍可能在MS新出现的免疫机制中发挥作用,这提示MS和Harding病在治疗方面可能存在某种潜在联系,是未来值得探索的领域[4]。
通过该特殊罕见病例我们认识到Leber遗传性视神经病变的复杂性,尤其合并多样的神经系统症状时,诊断难度增加,易延误治疗时机,需加强对该病的认识。目前该病药物治疗还处于对症治疗阶段,虽然在基因位点和分子生物方面尚有较大探索的空间,但仍是极具挑战性的,因为罕见疾病的治疗效果很难研究。
利益冲突申明 本研究无任何利益冲突
作者贡献声明 王江南:实施研究;采集数据;起草稿件。李雪景:实施研究;采集数据;对文章的知识性内容作批判性审阅;行政、技术或材料支持。樊朕宏:采集数据
猜你喜欢
体育科技文献通报(2022年4期)2022-10-21 03:21:04
中国现代医生(2022年21期)2022-08-22 03:30:42
基层中医药(2021年8期)2021-11-02 06:24:54
科学(2020年3期)2020-11-26 08:18:30
中医眼耳鼻喉杂志(2019年3期)2019-04-13 05:26:50
好日子(2018年9期)2018-10-12 09:57:20
中国体育教练员(2018年2期)2018-07-23 02:49:16
Coco薇(2017年12期)2018-01-03 21:18:37
中国医药科学(2017年9期)2017-08-04 21:42:14
放射学实践(2016年6期)2016-12-15 21:55:30
