
异唑虫酰胺合成工艺研究
2024-04-08 01:46:32刘安昌吴子豪陈典富
现代农药 2024年1期
刘安昌,吴子豪,陈典富,方 强
(武汉工程大学化工与制药学院,武汉 430074)

图1 异唑虫酰胺结构式
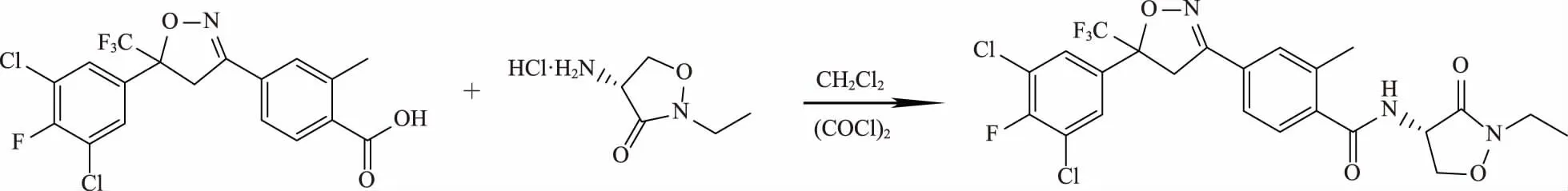
图2 异唑虫酰胺的合成路线
其中,中间体4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸主要有2条合成路线。
路线1:以4-乙酰基-2-甲基苯甲酸甲酯[4]为原料,在三乙胺作用下与3,5-二氯-4-氟三氟苯乙酮缩合,然后在2,6-二甲基吡啶的催化下与羟胺反应得到4-[5-(3,5-二氯-4-氟苯基)-4,4,4-三氟-3-羟基-1-(羟基亚氨基)丁基]-2-甲基苯甲酸甲酯;其在六甲基二硅氮烷锂盐(LiHMDS)和甲基磺酰氯的作用下环化、水解得到4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸。该工艺用到昂贵的LiHMDS,难于工业化。其合成路线见图3。
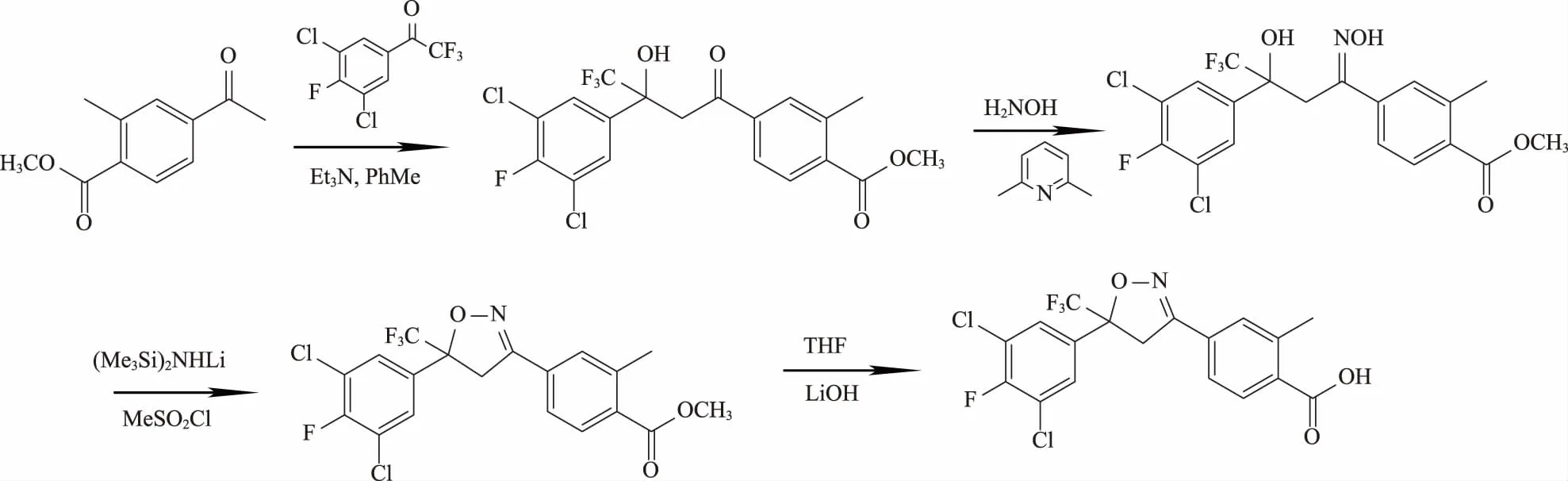
图3 4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸合成路线1
路线2:以1,3-二氯-2-氟-5-(3,3,3-三氟丙-1-烯-2-基)苯在三乙胺和碳酸钾的作用下与4-[(Z)-氯羟胺甲基]-2-甲基苯甲酸叔丁酯得到4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸叔丁酯[3],再水解得到4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸。该路线起始原料不易得,不易工业化。其合成路线如图4。
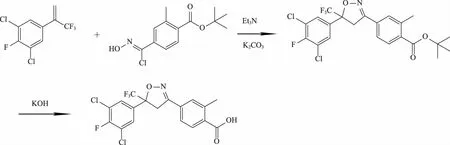
图4 4-[5-(3,5-二氯-4-氟-苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸合成路线2

图5 4-氨基-2-乙基-3-异唑烷酮盐酸盐合成路线
综上所述,采用3,5-二氯-4-氟溴苯为起始原料合成中间体4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸;以D-环丝氨酸为原料,制得4-氨基-2-乙基-3-异唑烷酮盐酸盐。两者经缩合反应合成异唑虫酰胺。该路线原料易得,反应温和,适应工业化。优化合成路线如图6。
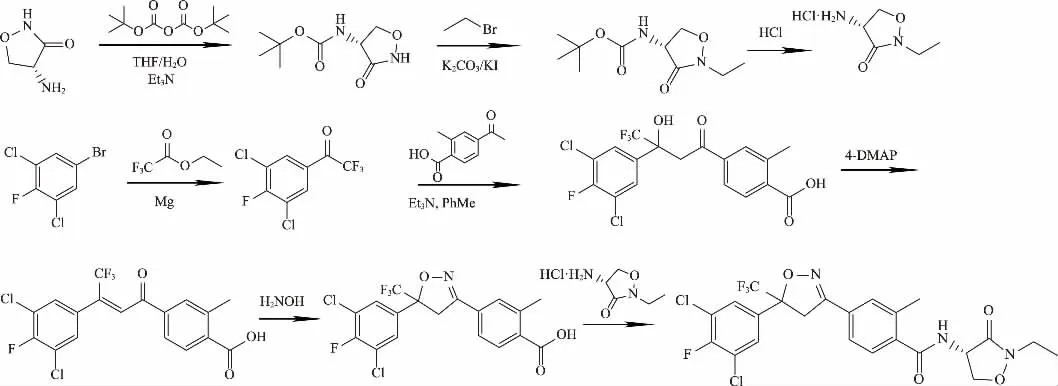
图6 异唑虫酰胺的优化合成路线
1 实验部分
1.1 实验材料
3,5-二氯-4-氟溴苯(≥99.0%),上海麦克林生化科技有限公司;2,2,2-三氟乙酸乙酯,上海升德医药科技有限公司;D-环丝氨酸(≥99.0%),上海笛柏生物科技有限公司;二碳酸二叔丁酯(99.0%),上海毕得医药科技股份有限公司;50%羟胺水溶液,上海凛恩科技发展有限公司;溴乙烷(98%),上海恩氟佳科技有限公司;其他试剂均为国药分析纯。
1.2 2,2,2-三氟-1-(3,5-二氯-4-氟苯基)乙酮的合成
在装有搅拌器和温度计的500 mL反应瓶中,加入150 mL干燥的四氢呋喃和5.04 g(0.21 mol)金属镁,加热至40~50℃,滴加48.6 g(0.2 mol)3,5-二氯-4-氟溴苯,待反应完全后,冷却至室温。向反应液中滴加56.8 g(0.4 mol)2,2,2-三氟乙酸乙酯,在室温下反应8 h。反应完全后,蒸出部分四氢呋喃,用50 mL二氯甲烷萃取2次,合并有机层,无水硫酸钠干燥,浓缩得黑色液体。减压蒸馏,收集2 mmHg时62~70℃的馏分,得36.5 g淡黄色液体,收率70.3%。1H NMR(CDCl3)δ:8.05(s,2H)。
1.3 4-[3-(3,5-二氯-4-氟苯基)-4,4,4-三氟-3-羟基-丁酰基]-2-甲基苯甲酸的合成
将17.8 g(0.1 mol)4-乙酰基-2-甲基苯甲酸、26.7 g(0.11 mol)2,2,2-三氟-1-(3,5-二氯-4-氟苯基)乙酮、150 mL甲苯和11.1 g(0.1 mol)三乙胺加入250 mL的反应瓶中,于50℃下搅拌20 h。反应液冷却至25℃,加入150 mL乙酸乙酯,用HCl水溶液调节pH至1~2,分出有机层,水层用60 mL乙酸乙酯萃取2次,合并有机相,用60 mL水洗2次。浓缩得白色微黄固体,加入正己烷搅拌结晶,过滤、干燥,得白色固体33.4 g,收率为76%。
1.4 4-[3-(3,5-二氯-4-氟苯基)-4,4,4-三氟-1-氧代-2-丁烯-1-基]-2-甲基苯甲酸的合成
将8.78 g(0.02 mol)4-[3-(3,5-二氯-4-氟苯基)-4,4,4-三氟-3-羟基-丁酰基]-2-甲基苯甲酸、0.35 g(0.003 mol)4-二甲氨基吡啶、60 mL甲苯加入250 mL的反应瓶中,于50℃下滴加5.1 g(0.05 mol)乙酸酐搅拌10 h。冷却,加入50 mL甲苯,用HCl水溶液调节pH至1~2,分出有机层,水层用50 mL甲苯萃取,合并有机相,用60 mL水洗2次。浓缩得黄色稠状固体8.1 g,加入正己烷搅拌结晶,过滤、干燥,得黄色固体7.8 g,收率为92.6%。
1.5 4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸的合成
将5 g(0.012 mol)4-[3-(3,5-二氯-4-氟苯基)-4,4,4-三氟-1-氧代-2-丁烯-1-基]-2-甲基苯甲酸、2.4 g(0.036 mol)50%羟胺水溶液、1.6 g(3.63 mmol)四丁基溴化铵依次加入装有80 mL二氯甲烷溶液的反应瓶中,冷却至-10℃。将1.6 g氢氧化钠固体溶于5 mL水中,配制氢氧化钠水溶液,冷却后缓慢滴加至反应瓶中,滴加完毕后保温8 h,随后升温至25℃反应过夜。反应结束后加100 mL水稀释,调节溶液pH至1~2,补加50 mL二氯甲烷充分搅拌30 min,静置分层,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤、浓缩得棕黄色油状物,加入正己烷+乙酸乙酯(体积比4∶1)混合溶剂重结晶,得浅黄色固体3.43 g,收率为65%。1H NMR(CDCl3)δ:2.70(s,3H)、3.74(d,1H)、4.12(d,1H)、7.54~7.62(m,4H)、8.11(m,1H)。
1.6 (R)-氨基甲酸(3-氧代-4-异唑烷基)-1,1-二甲基乙基酯的合成
将4.1 g(0.04 mol)D-环丝氨酸溶于50 mL四氢呋喃和50 mL水的混合溶液中,滴加4.44 g(0.044 mol)三乙胺,滴加完毕后溶液冷却至0℃。向其中滴加9.34 g(0.043 mol)二碳酸二叔丁酯的四氢呋喃(15 mL)溶液,30 min内滴加完毕并保温3 h,随后升至室温反应过夜。减压蒸馏除去溶剂后,缓慢滴加2 mol/L盐酸水溶液调节pH至2~3,出现大量白色固体。过滤、干燥得白色固体6.2 g,收率为76%。
1.7 (R)-氨基甲酸(2-乙基-3-氧代-4-异唑烷基)-1,1-二甲基乙基酯的合成
将5.05 g(0.025 mol)(R)-氨基甲酸(3-氧代-4-异唑烷基)-1,1-二甲基乙基酯溶于60 mL N,N-二甲基甲酰胺中,然后加入8.64 g(0.062 5 mol)碳酸钾、4.6 g(0.027 5 mol)碘化钾,混合物在室温下搅拌30 min。将反应混合物冷却至0℃,并在30 min内滴加3 g(0.027 5 mol)溴乙烷,反应液室温下搅拌过夜。真空除去溶剂,加入50 mL乙酸乙酯和80 mL水萃取,有机层用饱和食盐水洗涤,无水硫酸钠干燥,过滤后浓缩,得白色固体4.03 g,收率为70%。
1.8 4-氨基-2-乙基-3-异唑烷酮盐酸盐的合成
将5 g(0.022 mol)(R)-氨基甲酸(2-乙基-3-氧代-4-异唑烷基)-1,1-二甲基乙基酯溶于4 mol/L氯化氢的1,4-二氧六环溶液(100 mL)中,升温至35℃搅拌过夜,出现白色浑浊。将混合液冷却至0℃继续搅拌1 h,过滤得白色固体,滤饼用冰1,4-二氧六环冲洗,干燥得3.75 g白色固体,收率为65%。1H NMR(DMSO)δ:9.04(br,3H)、4.62(m,1H)、4.51(m,1H)、4.20(m,1H)、3.57(m,2H)、1.13(t,3H)。
1.9 异唑虫酰胺的合成
向装有6.34 g(0.015 mol)4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸的反应瓶中依次加入30 mL二氯甲烷、8 mL草酰氯和1滴N,N-二甲基甲酰胺,室温反应5 h。浓缩除去溶剂,将浓缩液溶于30 mL四氢呋喃中,室温下向其中滴加7 mL三乙胺。将3.12 g(0.018 5 mol)4-氨基-2-乙基-3-异唑烷酮盐酸盐溶于50 mL四氢呋喃中,并将其滴入反应瓶内,室温下反应过夜。反应结束后减压除去溶剂,加水稀释,并用乙酸乙酯萃取,有机层用饱和食盐水洗涤,无水硫酸钠干燥。过滤、浓缩,得到棕黄色粘稠固体,用正己烷重结晶,得黄色固体4.76 g,收率为58%。1H NMR(CDCl3)δ:7.60(d,2H)、7.55(s,1H)、7.51(s,2H)、6.43(d,1H)、4.94~5.05(m,1H)、4.86(dd,1H)、3.99~4.13(m,2H)、3.59~3.78(m,3H)、2.50(s,3H)、1.21~1.35(t,3H)。
2 结论
以3,5-二氯-4-氟溴苯为起始原料,经格氏反应与2,2,2-三氟乙酸乙酯反应得到2,2,2-三氟-1-(3,5-二氯-4-氟苯基)乙酮,然后与4-乙酰基-2-甲基苯甲酸经缩合、脱水、环化反应得到4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸。
以D-环丝氨酸为原料,与二碳酸二叔丁酯反应生成(R)-氨基甲酸(3-氧代-4-异唑烷基)-1,1-二甲基乙基酯,得到Boc保护基团。以碳酸钾为缚酸剂,在碘化钾的催化作用下,与溴乙烷反应,并在酸性条件下脱去Boc保护基,得到4-氨基-2-乙基-3-异唑烷酮盐酸盐。
4-[5-(3,5-二氯-4-氟苯基)-4,5-二氢-5-(三氟甲基)-3-异唑基]-2-甲基苯甲酸与草酰氯发生酰基化反应,再在三乙胺的作用下与4-氨基-2-乙基-3-异唑烷酮盐酸盐反应生成目标产物异唑虫酰胺。
猜你喜欢
山东化工(2024年1期)2024-02-04 09:47:12
云南化工(2021年10期)2021-12-21 07:33:24
中国资源综合利用(2017年4期)2018-01-22 02:46:55
中国资源综合利用(2016年7期)2016-02-03 03:00:19
合成化学(2015年10期)2016-01-17 08:56:30
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:14
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
有机氟工业(2014年3期)2014-06-05 14:36:38
火炸药学报(2014年5期)2014-03-20 13:17:51
影像科学与光化学(2014年5期)2014-03-11 16:03:04
