
镁基固态储氢材料的研究进展
2024-04-02 06:45梁宸曦王振斌张明锦马存花
储能科学与技术 2024年3期
梁宸曦,王振斌,2,张明锦,2,马存花,2,梁 宁
(1青海师范大学化学化工学院,青海 西宁 810016;2青海省人民政府-北京师范大学高原科学与可持续发展研究院,青海 西宁810008;3河南省地质矿产勘查开发局第三地质勘查院,河南郑州 451450;4河南省金属矿产深孔钻探工程技术研究中心,河南 郑州 450003)
20 世纪以来,煤、石油、天然气三大传统化石能源大规模消耗和大量温室气体的产生,促使科研人员对可替代能源进行了深入研究[1]。氢能作为一种理想的绿色能源,具有来源广泛、燃烧热值高(33.3 kWh/kg)、环境友好等优异特性,在可持续能源体系的发展与过渡阶段起着重要的作用[2]。氢可以从各种各样的过程中产生,如水的电解、化石燃料的等离子弧分解、水的热分解、生物质转化、光催化、生物光解等[3-7]。但氢气易燃易爆,且密度低,易逸出,对储存和运输带来不便。因此,储氢技术已成为当前氢能应用的关键步骤之一[8]。目前氢的存储方式主要包括以下几类:高压气态储氢[9]、液态储氢[10],以及固态储氢[11]。各种储氢方式的储氢原理和优缺点等具体细节见表1。
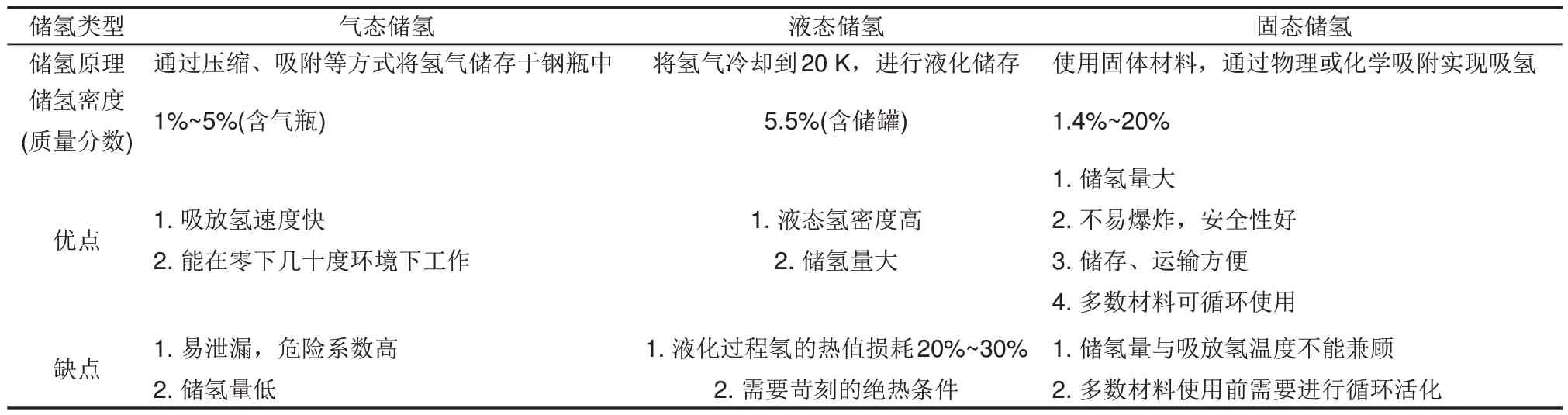
表1 气、液、固态储氢方式的比较Table 1 Comparison of gas, liquid and solid hydrogen storage methods
气态储氢可以储存约4%(质量分数)的氢气,但压缩过程充满着危险性,对容器耐压要求较高。液氢的密度为70.8 kg/m3,使用液态储氢系统可在一定程度上提高氢气储量,但液化过程中能耗高,对容器绝热性要求较高[12-13]。理想的储氢材料应具有高储氢密度、快速吸/放氢性能和长周期循环稳定性。国际能源署(International Energy Agency,IEA)提出理想储氢材料的性能标准为:质量储氢密度>5.5%(质量分数),放氢温度<423 K,循环寿命>1000 次[14-15]。与气态高压和液态储氢系统相比,固态储氢材料的储氢密度和安全性较高,具有满足IEA目标的巨大潜力。固态储氢材料根据吸附剂和吸附质之间作用力的不同可分为物理吸附和化学吸附两种方式。前者包括传统的碳基多孔材料、介孔材料、金属有机框架(MOFs)等。物理吸附主要通过相对较弱的范德华力实现储氢,吸附压强较高且只能在较低温度(77 K)下实现储氢[16]。与前者不同的是,化学吸附通过较强的化学键实现储氢,化学储氢材料包括化学氢化物(Mg-N-H、NH3BH3、Li-NH)[17-19]、络合氢化物(LiAlH4、LiBH4、NaBH4)[20-22]和金属氢化物(MgH2、TiFeH2、LaNi5H6)[23-25]等。尽管化学氢化物和络合氢化物普遍具有较高的储氢性能,但较高的工作温度和较差的循环可逆性限制了它们的大规模应用,此外,它们的合成和再生也是亟待解决的问题[26]。传统金属基氢化物(TiFeH2和LaNi5H6)可以在相对温和的操作条件下可逆地吸氢与脱氢,但由于金属含量较高,它们的质量储氢密度并不理想,例如,LaNi5的储氢量只有1.4%(质量分数)H2。因此,它们都不是固态储氢材料最为合适的选择。相比之下,MgH2的储氢性能优异(7.6%,质量分数)、无毒无害、可逆性好,而且具备氢净化功能,是一种极具前景的固态储氢材料[27-28]。但是,其较高的脱氢焓(ΔH=76 kJ/mol)以及脱氢活化能(Ea=160 kJ/mol)导致MgH2具有较高的脱氢温度[29-31]。此外,由于Mg—H键键能较高,H2分子的解离能力和H原子的重组能力较差,H原子在MgH2中扩散系数较低等问题的存在,设计一种能在适宜温度下快速实现吸脱氢的储氢材料仍然是一个挑战[32]。
本文首先介绍了MgH2的晶体结构和储氢机理,之后阐述了一系列改性方法,分析了不同改性方法所合成的MgH2储氢复合材料的性能特点,对其可逆吸脱氢量、热力学和动力学等性能进行了探讨。最后针对目前镁基固态储氢材料的研究进展以及未来的挑战进行了简要的总结与展望。
1 MgH2的晶格结构和储氢机理
为了改善MgH2的储氢性能,必须全面了解其结构稳定性。MgH2在不同的温度和压力条件下会以不同的晶体结构存在,分别是α-MgH2、β-MgH2、 γ -MgH2、 δ´ -MgH2、 ε -MgH2和cubic-MgH2。常见晶体结构及相应的压力-温度图(P-T图)如图1和图2所示[33]。已有研究证明MgH2的最低能态结构是具有四方晶系金红石型结构的α-MgH2[34-35]。MgH2的P-T图(图2)表明,α-MgH2会在2 GPa 的压力下转变为γ-MgH2。随着压力由2 GPa 进一步增加至7 GPa,MgH2的晶格结构会实现由γ-MgH2向ε-MgH2的转化。有报道称,通过高压扭转方法增加应变,α-MgH2可以完全转变为γ-MgH2,具有离子键的γ 相氢键结合能力较弱,可使脱氢温度降低80 K[36]。此外,在特定的压力和温度条件下,β-γ 和δ´-ε 有可能实现共存,不同结构类型的MgH2共存可能会对氢解吸性能产生催化作用。例如在γ-MgH2和β-MgH2的共存体系中,前者会诱导后者的Mg—H键失稳并释放氢气[37]。由于在相应温度和压力范围内,cubic-MgH2的吉布斯自由能高于其他五个相,因此cubic-MgH2没有出现在P-T图中。一系列现象证实了MgH2的吸脱氢性能与其晶体结构具有密切关系。
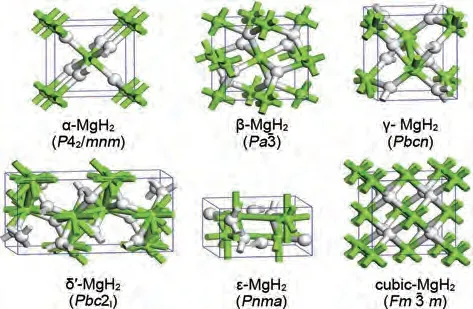
图1 MgH2的常见晶体结构[33]Fig.1 Common crystal structures of magnesium hydride [33]
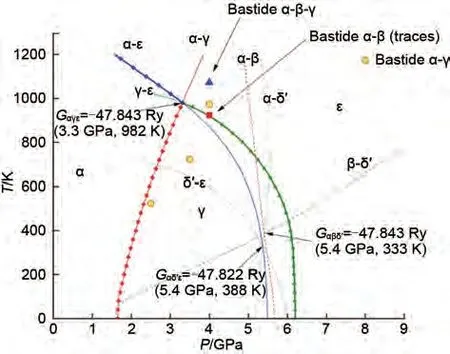
图2 MgH2的P-T图[33]Fig.2 The corresponding P-Tphase diagram of MgH2[33]
在阐述各种改性方法前需要对Mg/MgH2体系的储氢机理进行深入了解。Mg 在300~400 ℃,2.4~4 MPa条件下会与H2发生可逆化学反应从而实现吸脱氢,其反应方程式如式(1)所示:
正向为吸氢放热反应,逆向为脱氢吸热反应。如图3(a)所示,MgH2的吸氢反应由以下几个步骤组成[38]:①物理吸附,即镁通过范德华力将氢分子吸附至表面;②化学吸附,即解离的表面氢原子与镁形成Mg—H键;③固溶体α相的形成,即氢扩散到镁的晶胞空隙并发生占据;④固溶体β 相的形成,即晶胞中氢原子达到临界浓度,产生新的稳定相,PCT(压力-组成-温度)图上会出现一个平坦的平台,平台宽度代表着可逆储氢量;⑤β相生长和α 相的消失,平台逐渐消失,实现固溶体α 相到β相的完全转化。放氢反应是吸氢反应的逆过程。
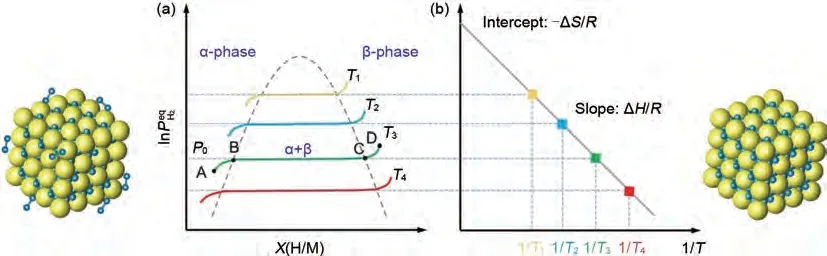
图3 (a) Mg/MgH2的压力成分等温线图[38];(b) Mg/MgH2相变相应的范特霍夫图[38]Fig.3 (a)Pressure component isotherm diagram of Mg/MgH2 and (b)Van´ t Hoff diagram corresponding to the Mg/MgH2 phase transition [38]
此外,通过测定不同温度下的PCT 曲线,氢化镁的脱氢焓(ΔH)和脱氢熵(ΔS)可通过Van't Hoff方程[图3(b)]进行计算,方程如式(2)所示:
其中,P0为大气压力(1.01×105Pa),ΔH和ΔS分别为MgH2的吸脱氢过程的焓变与熵变,T为吸放氢温度[图3(b)]。
2 MgH2的改性研究进展
2.1 合金化
合金化是一种改善镁基储氢材料吸脱氢性能的常用方法。常见合金储氢材料可分为稀土系(AB5型)、Zr系(AB2型)、Ti系(AB型)、镁系(A2B型)。储氢合金由两部分组成,一部分为吸氢元素或与氢有很强亲和力的元素A,它控制着储氢量的多少,是组成储氢合金的关键元素,主要是ⅠA~ⅤB 族金属,如Ti、Zr、Ca、Mg、V、Nb、稀土元素Re等;另一部分则为吸氢量较小或根本不吸氢的元素B,它则控制着吸/脱氢的可逆性,如Fe、Co、Ni、Cr、Cu、Al等[39-41]。而各体系储氢合金中,镁系合金的储氢质量密度最高,被学术界一致认为是目前最具应用潜力的储氢材料之一。典型的镁基合金体系是通过加入过渡金属元素或稀土元素与金属镁形成相应的合金,其中过渡金属元素可促进H2分解为H原子,从而降低氢解离和重组的Ea。此外,过渡金属未完全填充的空d电子轨道与氢原子相互作用会削弱Mg—H 键,从而降低储氢材料的脱氢Ea与ΔH,实现快速脱氢[42-45]。而稀土元素可形成分散或包覆Mg/MgH2的稀土氢化物抑制循环过程中出现的团聚现象,从而显著改善镁合金储氢材料的循环稳定性[46]。
镁基二元固态合金储氢材料的研究始于1967年,美国布鲁克-海文国家实验室的Reilly等[47]首次研究Mg-Cu 系储氢合金,但其仅有2.6%(质量分数)的理论储氢量,且Mg2Cu 和H2反应形成的MgCu2不参与吸氢。为了改善储氢性能,1968年,Reilly 等[48]通过混合熔炼首次制备了Mg2Ni 储氢合金,该合金理论储氢量提高至3.6%(质量分数)。此外,Mg2Ni合金还具有较好的热力学性能,Ni原子对H 的作用力强于Mg 原子对H 的作用,从而削弱了Mg—H键的相互作用。生成的Mg2NiH4二元氢化物的脱氢ΔH降至64 kJ/mol,相比MgH2的76 kJ/mol 显著降低,理论脱氢温度也由MgH2的300~400 ℃高温区间降至300 ℃以下。
鉴于Mg2NiH4合金优异的储氢性能,日本东北大学八木研究室[49]于1997 年首次采用氢化燃烧合成法(hydriding combustion synthesis,简称HCS法)直接合成了Mg2NiH4,该法合成过程在炉温低于870 K条件下进行,避免了镁的挥发,可直接从镁镍混合粉末制备出符合化学计量的镁镍储氢合金。由于反应中间产物Mg2Ni组织疏松、比表面积大和活性高的特点,一步法即可直接获得储氢合金氢化物[50]。HCS法对Mg-Ni储氢合金的制备方法进行了创新,也是对Reilly 等[47-48]的研究进行了补充与扩展,为Mg2NiH4储氢体系后续的快速发展与实际应用奠定了基础。
作为最具代表性的镁基储氢合金之一,Mg-Ni合金近年来也得到了更为深入的研究。Shao 等[51]机械球磨制备了Mg100-xNix系列合金,研究发现,Mg50Ni50合金表现出最佳的吸氢动力学和吸氢速率。该合金可以在373 K 下7 MPa 氢压下吸附1.85%(质量分数)H2(图4)。Tan等[52]在Shao等[51]的研究基础上,结合HSC 和机械球磨法合成了Mg100-xNix合金。差示扫描量热(DSC,图5)表明所有样品均显示两个脱氢峰,分别对应于β-MgH2和γ-MgH2。相比纯Mg,Mg100-xNix合金的峰值脱氢温度都有一定的下降。当Ni 添加量为20 %时,MgH2的初始/峰值脱氢温度从320.3 ℃/340.4 ℃降低到223.9 ℃/247.3 ℃。表明Ni 对解吸温度的降低有显著的作用。此外,473 K 下Mg100-xNix合金均表现出快速的氢化动力学,在100 s 内即可完成大于4%(质量分数)的饱和吸氢量。
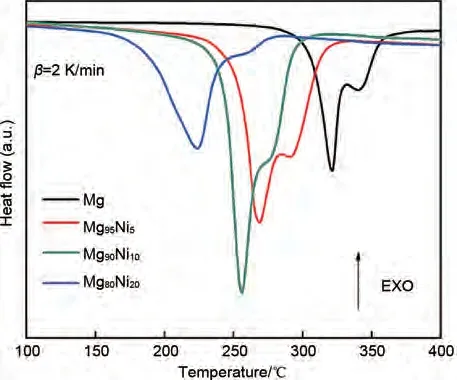
图5 HCS+MM后Mg100-xNix(x=0、5、10和20)合金的DSC曲线[52]Fig.5 DSC curves of Mg100-xNix(x=0, 5, 10 and 20)alloys after HCS+MM [52]
众所周知,空气中的H2O和O2容易与主要储氢元素Mg 反应从而降低镁合金的储氢性能。为了解决这一难题,朱云峰团队[53]将HCS 法制备的Mg80Ni20Hx暴露在空气中4个月后,研究了外部不利条件对Mg-Ni合金储氢性能的影响。研究发现,合成的Mg80Ni20Hx的脱氢峰值温度为387.77 ℃,并且随着空气暴露时间的延长而显著降低。在空气中暴露4 个月后,脱氢峰温度仅为239.83 ℃[图6(a)]。TPD 图[图6(b)]显示初始脱氢温度从344 ℃下降到215 ℃。图6(c)和(d)显示了不同温度下Mg80Ni20Hx在空气中暴露4个月后的等温吸/脱氢曲线。吸氢方面,在230 ℃、245 ℃、275 ℃和300 ℃下,接触空气后的储氢合金在400 s内吸收1.18%、2.59%、3.00%和3.04%(质量分数)H2。在300 ℃下,空气暴露后的Mg80Ni20Hx只需500 s即可实现饱和吸氢,远快于未接触空气的Mg80Ni20Hx(1500 s)。脱氢方面,在300 ℃下,接触空气后的储氢合金在400 s内脱附2.81%(质量分数)H2。在230 ℃、245 ℃和275 ℃下,分别在2500 s内脱附2.69%、2.80%和3.11%(质量分数)H2。在300 ℃下,空气暴露后的Mg80Ni20Hx可以在1000 s 内完全脱氢,远快于未接触空气的Mg80Ni20Hx(2500 s)。 经理论计算,Mg80Ni20Hx在空气中暴露4个月后,其脱氢活化能Ea为(63.56±2.43) kJ/mol,较未接触空气的Mg80Ni20Hx(93.5 kJ/mol[54])明显降低[图6(e)、(f)]。
除了典型的Mg-Ni储氢合金外,Al、Fe、Si以及镧系元素也可与Mg 形成稳定的Mg-Al 和Mg-稀土等二元合金相,在热力学上大幅降低Mg 基材料的脱氢焓。不同Mg 基储氢二元合金的储氢容量及脱氢焓如表2所示。
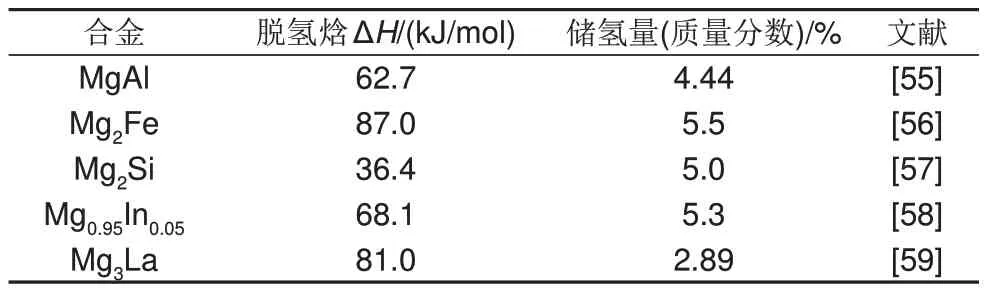
表2 一些二元镁合金的储氢量和脱氢焓Table 2 Hydrogen storage capacity and dehydrogenation enthalpy of some binary magnesium alloys
显然,一些金属元素的添加显著改善了镁基储氢合金的动力学性能和热力学性能,但是形成的二元镁基储氢合金的储氢量相较MgH2(7.6%,质量分数)显著降低。而当过渡元素、Al 等非过渡金属元素以及稀土元素复合添加时,得到的镁基复合材料的催化性能优于只添加其中一种,储氢量也有所提高。因此,为了兼顾高储氢量和优异的热力学/动力学性能,国内外学者对镁基储氢多元合金体系进行了深入探索,即向镁中同时添加两种或多种微量合金元素形成三元或高熵镁合金。
Wang 等[60]通过煅烧制备了Mg-Al 合金,并通过机械球磨法将过渡金属(TM=Ti、V、Ni)引入Mg-Al 合金中成功制备了Mg-Al-TM 三元合金。如图7(a)所示,Mg-Al合金的起始吸氢温度为188 ℃,在360 ℃时可逆储氢容量达到4.21%(质量分数),而Mg-Al-TM(TM=Ti、V、Ni)三元合金的起始吸氢温度为96 ℃,在360℃下的可逆储氢容量(质量分数)分别为4.28%、5.58%和5.77%。脱氢曲线[图7(b)]表明,当添加Ti、Ni和V后,Mg-Al合金的起始脱氢温度(310 ℃)显著降低。尤其是Mg-Al-V,其起始脱氢温度降至244 ℃(比Mg-Al 合金低66 ℃)。Mg-Al-V合金具有最可观的吸脱氢性能。
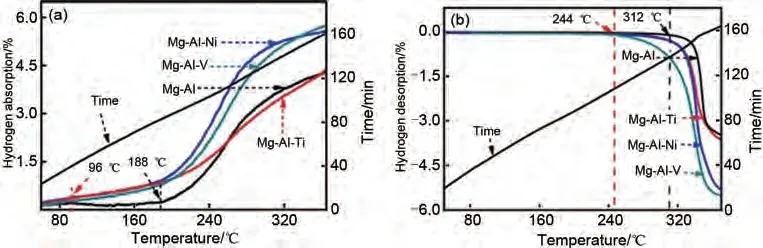
图7 Mg-Al和Mg-Al-TM(TM=Ti, Ni, V)合金的吸脱氢曲线图[60]Fig.7 Hydrogenation and dehydrogenation curves of the Mg-Al and Mg-Al-TM(TM=Ti、Ni、V)alloys [60]
Wei 等[61]通过真空感应熔炼法成功制备了Mg95-xAl5Yx(x=0~5)合金。通过线性拟合Arrhenius 方程发现:随着Y 含量由x=0 增加到5,该镁合金储氢材料的脱氢活化能从(155.562±2.29) kJ/mol 下降至(79.622±5.61) kJ/mol(图8),脱氢动力学性能得到显著改善。Mg-Al 基储氢合金的初始脱氢温度也随着Y 含量的增加降低了40.2 ℃,最大储氢量达到了5.1%(质量分数),显著高于Mg-Al 二元合金(4.44%,质量分数),兼顾了高储氢量和优异的热力学/动力学性能。该三元合金储氢性能的改善可归因于合金中不同元素之间产生的YH3催化相和成核活性位点。
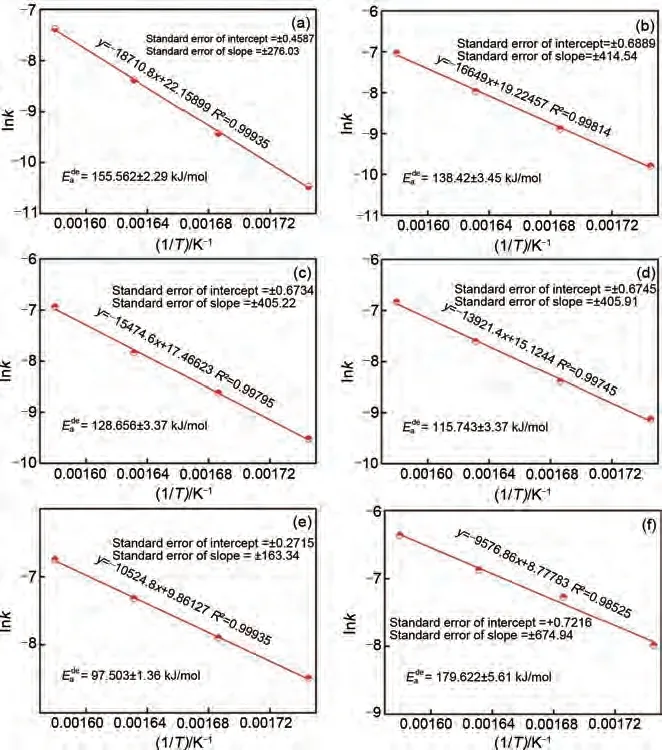
图8 Mg95-xAl5Yx(x=0~5)的阿仑尼乌斯方程[61]:(a)Y0 alloys; (b)Y1 alloys; (c)Y2 alloys; (d)Y3 alloys; (e)Y4 alloys; (f)Y5 alloysFig.8 Arrhenius plots of the Mg95-xAl5Yx(x=0~5)[61]: (a)Y0 alloys; (b)Y1 alloys; (c)Y2 alloys; (d)Y3 alloys; (e)Y4 alloys and (f)Y5 alloys
针对稀土镁合金储氢体系,目前研究最多的是Mg-Ni-稀土三元合金。前文提到了典型的Mg-Ni储氢二元合金的一些研究成果,而随着仪器设备和相关理论的完善,Mg-Ni合金的储氢温度也由300 ℃以下进一步降至250 ℃[62]。然而,Mg-Ni 二元合金的储氢性能依旧不够理想,仍有待进一步提高。针对以上问题,相关学者通过将部分稀土元素引入其中成功制备了一系列Mg-Ni-稀土三元合金体系,为高性能Mg-Ni系储氢合金成分设计与开发提供指导。
Yu 等[63]采用真空感应炉法制备Mg95-x-Nix-Y5(x=5, 10, 15)三元合金。通过对镍的组成、材料微观形态、储氢性能等进行系统研究后发现,YH3相在脱氢过程中不分解,均匀分散在母相合金中,对Mg/MgH2和Mg2Ni/Mg2NiH4的可逆相变起到了一定的催化和细化作用。通过密度泛函理论计算(density functional theory, DFT)发现,Ni 元素的加入可以有效降低MgH2的能带间隙,从而有效地改善镁基储氢合金的热力学性能[64],Ni5、Ni10和Ni15样品的脱氢焓分别降至84.5 kJ/mol、69.1 kJ/mol 和63.5 kJ/mol,Mg80Ni15Y5合金具有最佳的热力学性能,该合金初始吸氢温度也降至200 ℃。而吸脱氢过程中形成的YH3具有氢泵效应,使该三元储氢合金材料能够在1 min 内吸附约5.4%(质量分数)H2,达到了饱和储氢容量的90 %,显著改善了材料储氢性能(图9)。
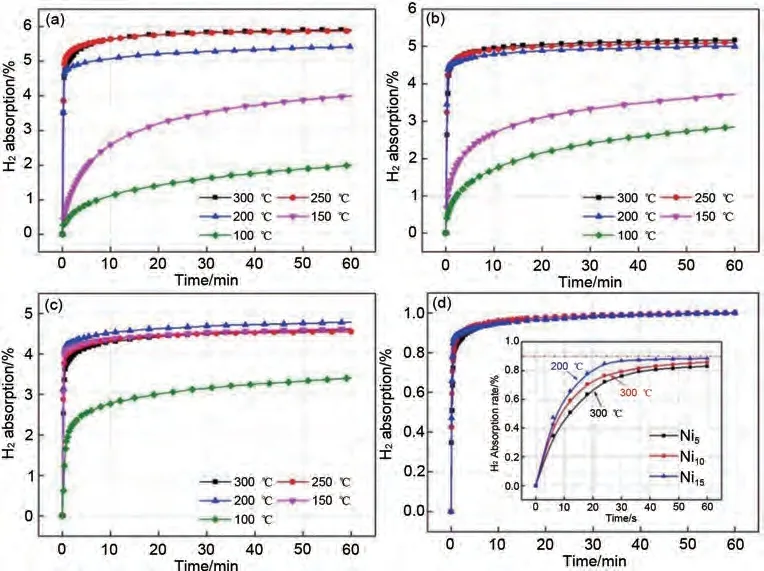
图9 活化后的Mg95-XNixY5(x=5,10,15)样品在不同温度下的等温吸氢曲线[63]:(a)Mg90Ni5Y5;(b)Mg85Ni10Y5;(c)Mg80Ni15Y5;(d)最佳氢化温度下各样品吸氢速率对比图Fig.9 Isothermal hydrogen absorption curves of the activated Mg95-xNixY5(x=5, 10, 15)samples in different temperatures: (a)Mg90Ni5Y5; (b)Mg85Ni10Y5; (c)Mg80Ni15Y5 and (d)the comparison of hydrogen absorption rate of these samples at optimum hydrogenated temperature [63]
为了了解镁基储氢合金的长期循环机理,Guo等[65]研究了Mg-Ni-La三元合金在623 K条件下经过200次循环后的循环可逆性。研究发现,Mg-Ni-La合金具有较好的脱氢循环性能,200次循环后脱氢量仍能保持在5.5%(质量分数)以上(图10)。随着循环次数的增加,样品的脱氢速率先增大后减小,最终达到相对稳定的状态。微观结构观察发现,在吸/脱氢过程中,由于体积的反复膨胀和收缩,纳米合金粉末同时存在烧结和氢爆两种现象。同时,原位形成的LaHx(x=2, 3)纳米晶体可在一定程度上抑制粉末的烧结。经过200次循环后,实验样品的平均粒径减小,比表面积明显增大,导致MgH2和Mg2NiH4的分解温度轻微向低温转移。此外,Mg2NiH4和LaHx(x=2, 3)在长期循环过程中被证明是稳定的催化剂,经过200次循环后仍能均匀分布在粉末中。
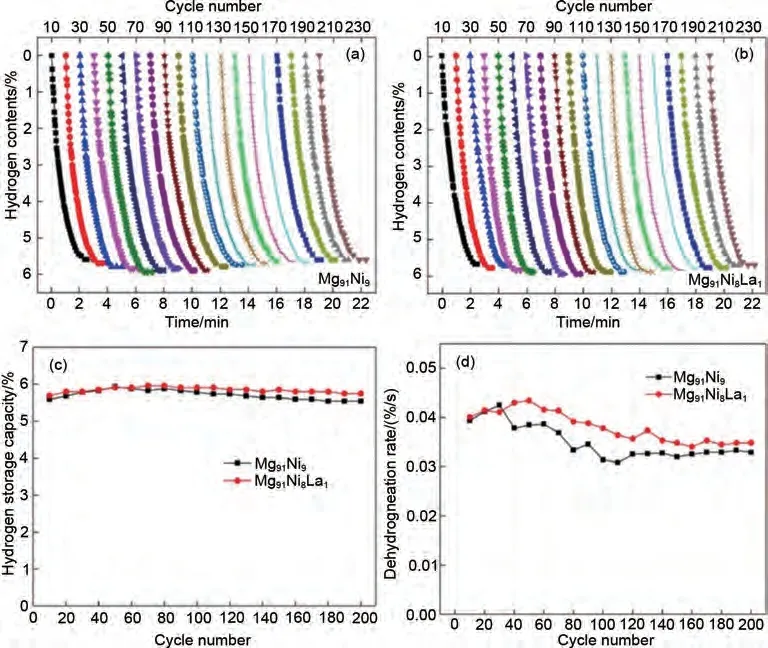
图10 Mg91Ni9与Mg91Ni8La1的200次循环的脱氢性能图[65]Fig.10 200-cycle dehydrogenation performance diagram of Mg91Ni9 and Mg91Ni8La1[65]
可以看出,三元储氢合金展现出更加优异的热力学与动力学性能,储氢量也有了明显的提高,近年来报道的一些Mg 基三元与四元合金相关储氢参数如表3所示。

表3 一些镁合金的相应储氢参数Table 3 Corresponding hydrogen storage parameters of some magnesium alloys
虽然前文提及的一系列三元镁合金展现出了比简单二元合金更加理想的储氢性能,但是总体来说,三元镁合金的脱氢温度和脱氢活化能等参数需要进一步改善。由此一种新型合金——高熵合金(HEA)进入了学者们的研究视野中。
高熵合金由中国台湾学者叶筠蔚等[74]于1995年首次提出,打破了传统合金固有的设计思路。高熵合金是指由不少于5种元素所组成的合金,各原子占比最大不超过35%最小不低于5%,而传统合金则含有一种原子百分占比大于50%的元素。近年来,将高熵镁合金作为储氢合金进行研究的相关报道还很少,但由于多个不同半径原子之间理化性质差异导致的严重晶格畸变为氢原子成核和扩散提供了更多的空间,这使得高熵镁合金成为了极具潜力的储氢合金。各国学者也开始对高熵储氢镁合金进行了深入的探索。
申炳泽等[75]通过机械球磨法制备了MgxTiVNiAlCr(x=0, 0.5, 1)。图11 为高熵合金TiVNiAlCr、Mg0.5TiVNiAlCr 以及MgTiVNiAlCr 分别在473 K、523 K 以及573 K 下的等温吸氢曲线。可以看出,各试样随着温度的升高吸氢量都会有一定程度的下降,因为大多数吸附都是放热过程,温度升高吸附量会降低,所以简单的升温来促进氢气的热扩散是不可取的。在473 K 温度条件下TiVNiAlCr、Mg0.5TiVNiAlCr 以及MgTiVNiAlCr 的最大储氢量(质量分数)分别为0.57%、0.7%、1.3%。储氢量随着Mg 含量的增加而显著改善,但是储氢量较低。总的来说,Mg 元素的添加对高熵合金的储氢性能有所提高,但仍无法达到理想的储氢量,有待进一步改善。
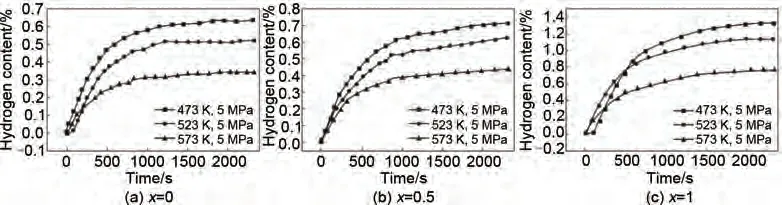
图11 高熵合金MgxTiVNiAlCr在473 K、523 K、573 K下的等温吸氢曲线[75]Fig.11 Hydrogen absorption curves of MgxTiVNiAlCr(x=0, 0.5, 1)at 473 K, 523 K, 573 K [75]
为了进一步提高含镁高熵合金的储氢量,Montero 等[76]在惰性气氛下,采用机械化学合成方法制备了一种新型高熵合金Mg0.1Ti0.3V0.25Zr0.1Nb0.25。该合金在25 ℃室温下迅速吸氢,在1 分钟内达到1.72H/M(H/M 指单位质量材料的储氢能量)的最大吸氢量,吸氢速率与吸氢量相比上文提到的MgTiVNiAlCr(1.3%,质量分数)都有了明显的改善。经过一次吸脱氢循环后,Mg0.1Ti0.3V0.25Zr0.1Nb0.25合金的储氢量有略微下降。吸脱氢循环性能测试(图12)表明Mg0.1Ti0.3V0.25Zr0.1Nb0.25五元合金在第二个循环中储氢容量损失了约11%(质量分数从2.7%降到2.41%),随后几个循环的可逆容量稳定在2.4%左右。这与未添加镁元素的Ti0.325V0.275Zr0.125Nb0.275四元合金形成鲜明对比,Ti0.325V0.275Zr0.125Nb0.275在前4 次循环中损失近28%的储氢容量,然后达到约1.9%(质量分数)的稳定值。显然,含Mg 的HEA 比四元HEA(不含Mg)具有更大的可逆储氢量。
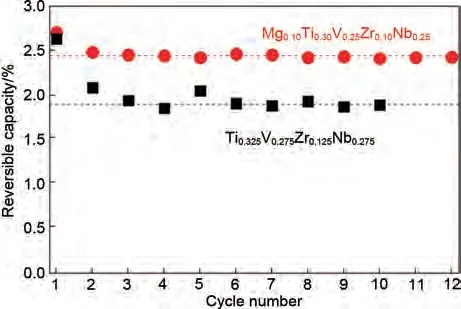
图12 Mg0.1Ti0.3V0.25Zr0.1Nb0.25和Ti0.325V0.275Zr0.125Nb0.275合金循环过程中可逆储氢容量对比图[76]Fig.12 Comparison between the variation of the reversible hydrogen storage capacity during cycling for the quinary Mg0.1Ti0.3V0.25Zr0.1Nb0.25 and quaternary Ti0.325V0.275Zr0.125Nb0.275 alloys [76]
本节具体概述了镁基二元、三元、四元以及高熵储氢合金各自的优缺点和发展脉络。总的来说,合金化可以有效降低脱氢温度以及脱氢焓变等参数,同时兼顾稳定的循环吸脱氢性能。但是在合金化过程中由于引入大量金属元素导致了储氢合金氢容量的大幅下降。
2.2 纳米化
纳米化是同时调节MgH2热力学和动力学的改性策略。由于纳米材料的量子尺寸效应、小尺寸效应和表面效应,与大体积MgH2相比,纳米MgH2具有更高的氢扩散系数以及良好的吸/脱氢动力学。理论计算表明,当晶粒尺寸在11~1.3 nm时,氢解吸活化能显著降低。当粒径降至0.9 nm 时,解吸温度下降到200 ℃[77-79]。已报道的制备纳米镁基材料的纳米技术包括高能球磨法[80]、化学还原法[81]、气相沉积法[82]、纳米限域法[83]等。
2.2.1 高能球磨法
高能球磨(HEBM)是制备镁纳米颗粒广泛使用的方法之一,球磨过程形成了不稳定的γ-MgH2相、应力、应变、晶体缺陷以及大量的纳米晶界和相界,可在一定程度上改善MgH2的储氢性能。Schulz等[84]首次发现高能球磨可以获得具有高比表面积的新型纳米结构,在300 ℃下400 s内吸收7%(质量分数)H2,在350 ℃下600 s内脱附7%(质量分数)H2。结果表明,机械球磨引起的结构缺陷降低了吸/脱氢的活化能。Varin等[85]进一步研究了MgH2的平均粒径随不同研磨时间的变化,发现当粒径减小到600 nm时,脱氢温度比纯MgH2低60 ℃。
然而高能球磨法易引入杂质颗粒,容易分布不均,并且易引起材料的团聚和长大,使得循环稳定性降低。
介质阻挡放电等离子体辅助球磨(DBDP)是Ouyang 等[86]发展的一种改进球磨技术。由于材料不仅受到机械球磨机的粉碎和冲击,而且还受到高能非平衡等离子体的热冲击和活化效应,DBDP的研磨效率明显高于HEBM。Dan等[87]通过等离子体球磨工艺在MgH2表面合成了超细镍纳米粒子(2~6 nm)。复合材料在275 ℃下10分钟内可快速脱附超过6.5%(质量分数)H2,并且在九个吸/脱氢循环中几乎没有容量衰减(图13)。
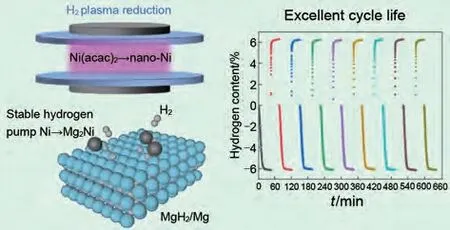
图13 纳米MgH2-Ni体系的催化机理图及循环性能图[87]Fig.13 Schematic of the catalytic mechanism and cycle properties of the nano-MgH2-Ni system [87]
DBDP为制备纳米材料和提高球磨效率提供了一种可行、低成本、无污染的方法,对先进镁基储氢材料的设计和制备具有重要的理论指导意义。
氢化燃烧合成(HCS)以其活性高、无活化产物以及产物吸氢速率快而著称,相关科研人员结合HCS 与HEBM 进行了广泛的研究。Liu 等[88]结合氢化燃烧合成和高能研磨(HCS+HEBM)制备了纳米结构化的Mg2Ni,发现吸氢速率在313 K 和373 K明显增加。初始脱氢温度为370 K,比HCS产物低约190 K。最近,韩树民团队[89]通过HCS+MM 将MgC0.5Co3化合物引入MgH2。材料在325 ℃条件下60 min 内脱氢量达到4.38%(质量分数),接近纯MgH2的4.5 倍。此外,解吸活化能从纯MgH2的(162.8±8.3) kJ/mol 降 低 到(126.7±1.4) kJ/mol。MgC0.5Co3保持相对稳定,循环后没有发生任何化学转化(图14)。
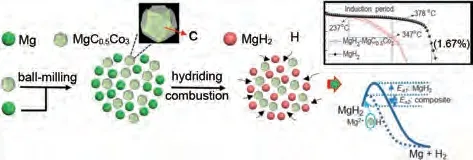
图14 MgC0.5Co3催化机理图[89]Fig.14 Diagram of the catalytic mechanism of MgC0.5Co3[89]
2.2.2 化学还原法
化学还原是合成Mg/MgH2纳米颗粒的有效方法。Rieke等[90]于1972年首次通过碱金属还原镁盐成功获得了高活性镁粉。之后,双环戊二烯基镁(MgCp2)、二正丁基镁(MgBu2)和氯化镁等也被选为Mg源得到了广泛研究[91-93]。化学还原通常会引入电子载流子来加速电子在碱金属和镁盐之间的转移过程。由于镁的低氧化还原电位(EMg2+/Mg=-2.37 V),镁盐在非水溶液的非质子溶剂中被电化学还原,整个体系由作为电解质的表面活性剂以及纳米颗粒稳定剂组成。2018 年,Shen 等[94]以TBAB 为表面活性剂,通过对Mg(BH4)2的电化学还原,获得了Mg 20纳米颗粒(60~100 nm)。Mg纳米颗粒在100 ℃下氢化后,完全转化为β-MgH2(70.4 %)和γ-MgH2(29.6 %)。γ-MgH2在一个循环后稳定存在,γ-MgH2的存在使得脱氢活化能下降到(69.1±2.9) kJ/mol,脱氢焓下降至(57.5±5.3) kJ/mol。Jeon 等[95]报道了在含有锂、萘和聚甲基丙烯酸甲酯(PMMA)的四氢呋喃溶液中还原二茂镁(MgCp2)的方法,获得了能隔绝水氧的聚合物基纳米Mg 复合材料(Mg NCs/PMMA),Mg NCs 粒径约为5 nm。在200 ℃下30 min内可以快速吸附4%(质量分数)H2,PMMA有效抑制了循环过程中Mg/MgH2的团聚与粗化(图15)。Francois 等[96]以溴化四丁胺(TBA)为模板在四氢呋喃中电化学还原制备了5 nm的Mg纳米胶体,该材料在室温下能够可逆储存7.6%(质量分数)H2,在85 ℃下完全脱氢。Norberg等[97]在联苯、苯或萘为电子载体的情况下,用钾(K)还原MgCp2制备了25~38 nm的镁单晶,发现25 nmMg纳米晶体表现出最好的吸/脱氢性能。
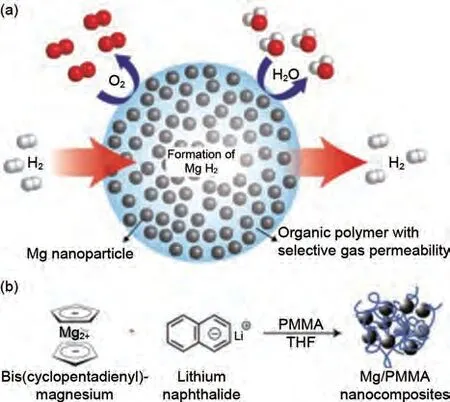
图15 Mg NCs/PMMA在空气暴露条件下的储氢机理图[95]Fig.15 Diagram of the mechanism of hydrogen storage under air exposure of Mg NCs/PMMA [95]
然而,化学还原法难以控制颗粒的规模和形态,合成的镁颗粒的形貌无明显差异。此外,萘、联苯或苯等电子载体会保留在储氢材料中,一定程度上抑制储氢性能。
2.2.3 气相沉积法
除了高能球磨、化学还原法外,气相沉积也是一种常见的纳米改性方法。气相沉积法是基于电弧产生高温使金属瞬间蒸发,在氢气等气体作用下使金属原子经历蒸发、形核、长大、凝聚等一系列过程,以获得纳米Mg。例如,Matsumoto 等[98]通过调节温度和氢压,利用化学气相沉积(CVD)成功制备了三种形貌的MgH2。Li 等[99]通过物理气相沉积(PVD)合成了直径为30~50 nm、80~100 nm 和150~170 nm 的Mg 纳米线,发现它们的脱氢活化能和脱氢焓变随着纳米线直径的减小而减小。直径最小(30 nm)的Mg 纳米线在300 ℃下30 min 内吸收7.6%H2并释放6.8%H2。后来,该课题组[99]构建了直径更小的Mg 纳米线理论模型,当纳米线直径只有0.85 nm 时,脱氢焓只有34.5 kJ/mol。但是,到目前为止,这种尺寸的纳米线还难以通过实验合成。Lu 等[100]采用气相沉积法制备了核壳状的Mg@Pt纳米颗粒(约200 nm)。Mg3Pt充当MgH2放氢的“氢泵”,改善了吸脱氢动力学,初始脱氢温度由MgH2的333.3 ℃降至287.5 ℃。但脱氢温度仍然难以满足实际应用条件,有待进一步改善。
此外,气相沉积法还可用于制造多层纳米级Mg/MgH2薄膜[101]。Higuchi 等[102]通过射频磁控溅射设计了Pd(50 nm)/Mg(xnm)/Pd(50 nm)三明治结构薄膜(图16)。脱氢温度降低至100 ℃以下。薄膜中的压缩应力和PdH2对MgH2的氢泵效应导致了脱氢温度的下降。

图16 Pd (25 nm)/Mg (200 nm)(a)和Pd (50 nm)/Mg(200 nm)/Pd (50 nm)(b)薄膜截面的TEM图[101]Fig.16 TEM micrographs for the cross section of (a)Pd (25 nm)/Mg (200 nm) and (b) Pd (50 nm)/Mg (200 nm)/Pd (50 nm) films [101]
多层膜结构有利于研究相应的催化机理。其组成、界面和结晶度可以在纳米尺度上精确调整,但制备过程复杂且材料昂贵。需要进一步开发适合实际生产条件的沉积方法。
2.2.4 纳米限域法
纳米限域(简称NC)是将Mg/MgH2纳米团簇限制在多孔支架中,多孔支架作为纳米材料的团聚抑制剂和尺寸控制剂显著改善了镁基材料的储氢性能。石墨烯和碳纳米管等碳材料作为支架已广泛应用于MgH2的改性研究[103-104]。Xia 等[105]在石墨烯的结构导向效应下,通过加氢反应诱导自组装制备了均匀分布的单分散MgH2纳米颗粒,该材料在150 ℃下7 min内可吸附5.4%(质量分数)H2。MgH2与石墨烯之间的强耦合保证了纳米结构的稳定性。此外,石墨烯的高热导率可以促进热扩散,提高氢气的吸附/脱附速率,从而获得了优异的性能。除碳基材料外,聚甲基丙烯酸甲酯(PMMA)、金属-有机框架(MOFs)以及介孔CoS纳米盒(CoS-NB)作为支架材料也得到了广泛研究。Ruminski 等[106]将不同比例的Mg纳米颗粒包覆在PMMA中,研究了包覆聚合物对纳米晶镁的空气稳定性和储氢密度的影响(图17)。含65%(质量分数)Mg-PMMA 复合材料在空气中吸收了6.95%(质量分数)H2,在空气中暴露3 个月后几乎没有氧化,而含33.2%(质量分数)Mg-PMMA复合材料仅吸收了4.86%(质量分数)H2,在空气中完全氧化。研究表明,聚合物用量的减少提高了Mg-PMMA复合材料的空气稳定性和吸氢能力。Lim 等[107]通过对MOF 中双环戊二烯基镁(MgCp2)的热分解,制备了嵌在MOF 中的Mg 纳米晶体。它具有物理吸附和化学吸附的双重特性,表现出降低化学吸附/脱附温度的协同效应。邹建新团队[108]通过氢化浸渍在Ni-MOF 中的MgBu2,制备了尺寸约为3 nm的MgH2颗粒,研究表明,MgH2@Ni-MOF复合材料中Mg/MgH2的热力学[吸附/脱附焓分别为(65.7±2.1) kJ/mol 和(69.7±2.7) kJ/mol]和动力学[吸附/脱附活化能分别为(41.5±3.7) kJ/mol 和(144.7±7.8) kJ/mol]均显著改善。MgH2@Ni-MOF在热力学和动力学方面的改进归因于纳米约束的Mg/MgH2的“纳米效应”和原位形成的Mg2Ni/Mg2NiH4的催化效应。之后其团队[109]又通过模板法制备了限域在CoS-纳米盒(CoS-NBs)支架中的MgH2@CoS-NBs。MgH2@CoS-NBs 复 合 材 料 在548 K下2小时内脱附1.76%(质量分数)H2。即使在较低的温度(498 K)下,复合材料仍然可以在2小时内脱附1.13%(质量分数)H2。相比之下,纯MgH2样品在548 K 和498 K 下2 小时内只能脱附0.42%和0.16%(质量分数)H2。该复合材料经过10次的吸/脱氢循环后,吸氢动力学和吸氢能力方面没有明显的降低迹象,MgH2@CoS-NBs 复合材料优异的吸氢动力学和循环稳定性主要归因于纳米限制的Mg/MgH2晶体的“纳米尺寸效应”、MgS的催化作用和CoS-NBs支架的多功能作用。MgH2@CoS-NBs复合材料的具体制备过程与催化机理以及循环吸脱氢性能见图18。毫无疑问,纳米支架结构通过将其颗粒尺寸限制在纳米范围内,缩短了氢气的扩散路径,大大提高了MgH2的储氢热力学/动力学性能。然而,目前尚不清楚所观察到的增强效果是由于自身的限域性,还是由于氢化物与支架之间的化学相互作用、氢化物-溶剂加合物的形成等其他因素导致的。此外,体系储氢能力差的问题阻碍了这种方法在大规模储氢系统制备中的应用。
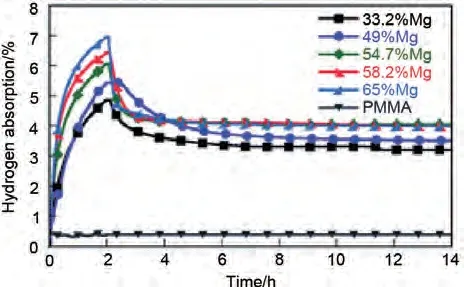
图17 Mg-PMMA 纳米复合材料的吸氢性能图[106]Fig.17 Hydrogen absorption of Mg-PMMA nanocomposites containing 33.2%, 49%, 54.7%,58.2% and 65% Mg [106]
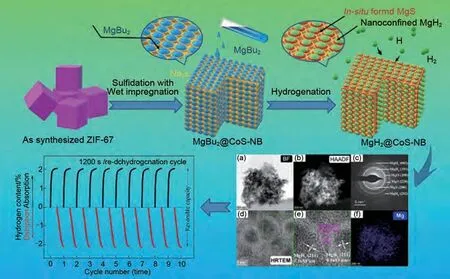
图18 MgH2@CoS-NBs的制备过程、催化相表征和循环性能图[109]Fig.18 Preparation process, catalytic phase characterization, and cycling performance diagram of MgH2@Co S-NBs [109]
本节就实现纳米化的各种制备方法进行了详细介绍,为了更直观地比较不同方法制备的纳米Mg/MgH2储氢性能方面的差异,表4对其进行了总结。
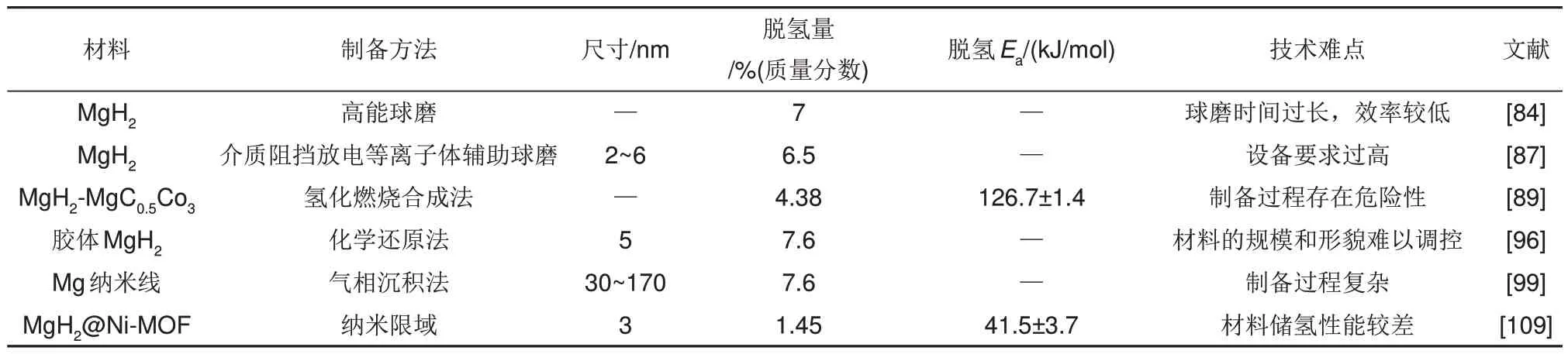
表4 不同方法制备的MgH2的性能差异和技术难点Table 4 Performance differences and technical difficulties of MgH2 prepared by different methods
2.3 催化剂改性
引入催化剂可以通过降低活化能,在一定程度上改善MgH2的吸/脱氢动力学。在过去的几年中,众多催化剂得到了广泛的探索和研究,包括金属、金属氧化物、其他金属化合物、金属与碳基复合催化剂、高熵合金等。本节将分别阐述不同种类的催化剂对镁基材料储氢性能的影响和作用机制。
2.3.1 过渡金属改性
研究发现[110-111],过渡金属往往有很多空轨道和占据轨道(轨道上有大量的d 电子),可以用来进行电荷转移、成键,因此过渡金属对氢原子具有很强的亲和力。在氢分子的解离或者氢原子的重组过程中,过渡金属的d电子和氢原子/氢分子轨道上的电子发生转移填充,由此产生的相互作用力可大幅降低氢分子解离或者氢原子重组的活化能,从而改善镁基材料的吸脱氢性能。过渡金属催化剂可分为Ti、Ni、Fe三大类。
针对过渡金属Ti,在1999年Liang等[112]用机械球磨法制备了MgH2-TM(TM=Ti、F、Mn、Fe、Ni)纳米复合材料。研究得出,含Ti的复合材料表现出最佳的解吸动力学,MgH2-5%(原子分数)Ti 复合材料在250 ℃下可在1000 s 内完全脱氢。Pukazhselvan 等[113]深入探讨了Ti 的催化机理,发现在Ti/MgH2纳米复合材料中通过强机械球磨可以形成稳定的TiH2-x。在脱/再氢化过程中,TiH2-x会转化为TiH2,使MgH2的脱氢活化能降低到89.4 kJ/mol,微观分析表明,TiH2晶体的尺寸约为Ti 晶体的4~5倍,表明催化剂引入引起的晶格应变是改变MgH2储氢性能的重要因素。
相比于简单球磨制备的MgH2-Ti 储氢材料,许多学者还使用一些新的制备方法将金属Ti直接引入Mg 当中,成功制备了具有优异吸脱氢性能的储氢材料。
Choi 等[114]采用化学蒸气合成(CVS)法制备Mg-5%(质量分数)Ti纳米粉体混合物,该方法一定程度上抑制了材料的团聚现象(图19),Mg-Ti 粉末几乎是球形的,直径为50~500 nm,只有小部分颗粒发生了轻微的团聚。图20 热重分析(TGA)表明CVS法制备的吸氢态MgH2-5%(质量分数)Ti纳米混合物(曲线A)的脱氢温度降至190 ℃左右,分别比商用MgH2(曲线B)和Ti球磨引入的MgH2(曲线C)的起始温度低约191 ℃和88 ℃。Mg-Ti 纳米粉末在70 分钟内实现完全脱氢。而商用MgH2和Ti 球磨引入的MgH2需要124 分钟以上。三个样品的起始脱氢温度和完全脱氢时间存在显著差异,表明与商用MgH2和Ti球磨引入的MgH2相比,通过化学蒸气合成(CVS)法制备的Mg-5%(质量分数)Ti 纳米材料的脱氢性能有了进一步改善,也证明了探究不同制样方法来改善材料性能的重要性。

图19 镁钛纳米粉末的TEM图[114]Fig.19 TEM image of the Mg-Ti nanopowder [114]
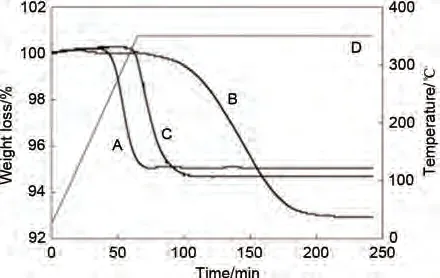
图20 Mg-Ti纳米粉末的氢化产物(A)MgH2-5%(质量分数)Ti(CVS)、(B)商用MgH2、(C)MgH2-5%(质量分数)Ti(球磨)的TGA曲线 [114]Fig.20 TGA profiles of the hydrogenated products of the Mg-Ti nanopowder(A)MgH2-5%Ti(CVS); (B)commerical MgH2; (C)MgH2-5%Ti(milled)[114]
针对过渡金属Ni,早在2005 年Hanada 等[115]就通过球磨将商用MgH2粉末与金属Ni混合,得到MgH2-Ni复合材料。如图21所示,通过热解吸质谱(TDMS)发现球磨2 小时后的Ni 引入复合材料的脱氢温度降至260 ℃,远低于纯MgH2(370 ℃)。球磨1小时导致起始脱氢温度降低至约250 ℃,而球磨15分钟导致峰值温度进一步降低至约210℃。由于球磨时间过短导致MgH2与Ni混合不均匀,峰值信号较弱。但通过控制球磨时间,Ni展现出了优异的催化效果。
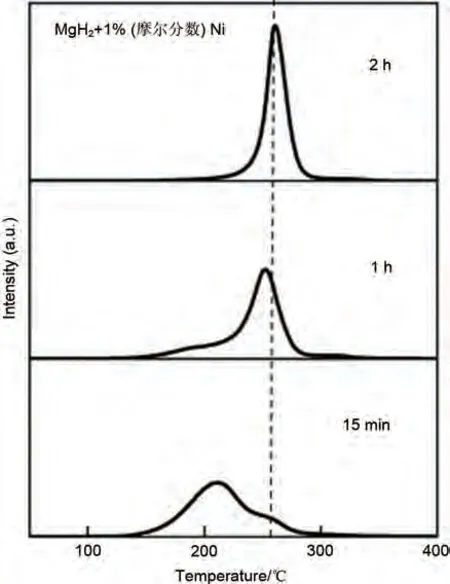
图21 1 MPa氢气气氛400 r/min下研磨15 min、1 h、2 h的MgH2-1%(摩尔分数)Ni的热解吸质谱[115]Fig.21 Thermal desorption mass spectra of hydrogen from the MgH2 composite with 1 mol% Ni by milling for 15 min, 1 h, and 2 h at 400 r/min under a hydrogen gas atmosphere of 1 MPa [115]
Ni 纳米粒子的优异催化作用得到证实。近年来,催化剂用量和颗粒尺寸等其他因素也得到了广泛研究。Xie等[116]研究了引入不同含量Ni纳米颗粒的MgH2的储氢动力学。等温脱氢曲线(见图22)表明,MgH2+10%(质量分数)纳米Ni 复合材料在250 ℃下可在10 min 内脱附6.1%H2。随着催化剂用量的增加,MgH2+纳米Ni复合材料的脱氢速率明显提高,但饱和储氢量会有一定衰减。结果表明,通过改变催化剂用量,可以进一步调控Ni 的催化作用。

图22 (a) 球磨MgH2;(b) MgH2+10%Ni;(c) MgH2+25%Ni;(d) MgH2+50%Ni和(e) MgH2+80%Ni在523 K时的等温脱氢曲线[116]Fig.22 Hydrogen desorption curves of the (a) MgH2 milled for 2 h; (b) MgH2+10%Ni; (c) MgH2+25% Ni;(d) MgH2+50% Ni, and (e) MgH2+80% Ni at 523 K under an initial pressure of 0.01bar of H2[116]
Yang 等[117]研究了Ni 颗粒尺寸对MgH2氢解吸性能的影响。结果表明,仅有2%(原子分数)细镍颗粒引入的MgH2在200 ℃时即可快速脱氢,当温度升至340 ℃时,脱氢量高达6.5%。然而,DSC曲线(见图23)表明,MgH2-2%(原子分数)Ni90(90 为Ni颗粒粒径)复合材料的峰值脱氢温度约为280 ℃,仅比MgH2-2%(原子分数)Ni200和MgH2-2%(原子分数)Ni100复合材料的峰值温度低约10 ℃。具有8%(原子分数)微尺寸Ni 的MgH2粉末在310 ℃和390 ℃下分两步释放氢气:第二步的脱氢温度(390 ℃)非常接近研磨态MgH2。因此,第二个吸热峰的存在可能是由于Ni 的不均匀分布使一些MgH2颗粒没有被完全催化导致的。即催化剂尺寸的减小并不是改善储氢材料性能的关键,催化剂在MgH2颗粒上的位点分布和密度才是提高MgH2吸脱氢性能的关键因素。

图23 简单球磨20 h的MgH2和不同类型的Ni(90 nm、100 nm、200 nm、7 μm)的DSC曲线[117]Fig.23 Comparison of the effects of different Ni particle sizes on the desorption properties of pre-milled MgH2 by simply mixed 20 h-pre-milled MgH2 and different types Ni (90 nm, 100 nm, 200 nm and 7 μm)[117]
针对过渡金属Fe,Fe是地球上最重要的元素,在自然界中广泛分布。近年来,其优异的催化性能受到了学者们的广泛关注。Bassetti 等[118]采用高能球磨法制备了MgH2-Fe 纳米复合材料。对不同Fe含量的MgH2进行球磨后发现,微晶尺寸、粉末粒径、催化剂浓度和分散性是影响脱氢动力学的关键因素。DSC 曲线分析表明,MgH2-Fe 的脱氢温度基本在220 ℃左右。添加10%(质量分数)Fe 时在300 ℃下、600 s 内释放约5%(质量分数)H2,具有最佳的催化效果。
众所周知,Fe原子可以通过吸附和取代Mg来提高储氢性能,但其潜在机理尚不清楚。为了解决此问题,Chen 等[119]基于密度泛函理论计算研究了Fe 原子不同引入形式对Mg 晶体性质的影响(图24)。通过模拟Fe 原子的吸附、解离和扩散过程,发现Fe原子能增强氢分子在Mg(0001)表面的吸附性能。氢解离计算表明,Fe 对H2解离的催化作用可以归结为被吸附Fe 原子的桥接作用,使电子从H2成键轨道转移到反成键轨道,从而削弱了H—H键。为了更好地了解Fe对Mg基体系氢吸附动力学的影响,Antiqueira等[120]采用10 h和24 h高能球磨技术对Mg-8%(质量分数)Fe 纳米复合材料进行了研磨。这两种纳米复合材料都具有非常快的吸收/解吸动力学。在350 ℃时,2 min内即可完全脱氢,10 min 内实现饱和吸氢。实验结果进一步表明,当磨粉时间延长至24 h 时,出现了Mg2FeH6与MgH2含量较高的混合物,Mg2FeH6的存在降低了复合材料的储氢能力,使解吸温度略有上升。因此,控制球磨时间是改善储氢材料性能的关键。
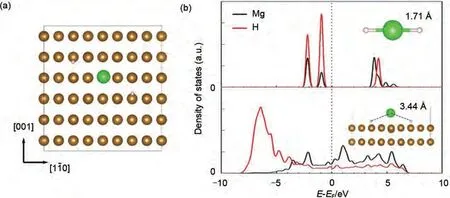
图24 MgH2在Fe(110)晶面上的吸附几何形状(棕色、绿色和白色球分别代表铁、镁和氢)(a);Mg和H在Fe(110)晶面上吸附之前(上)和吸附后(下)的预测态密度(PDOS)(b)[108]Fig.24 The adsorption geometry of MgH2 on Fe (110)surface in top view (a); The brown, green, and white ball represent Fe, Mg, and H, respectively, The projected density of states (PDOS)of Mg and H of MgH2 before (upper)and after (lower)adsorption on Fe (110)surface (b) [108]
Zhang等[121]采用湿法化学球磨法制备了厚度约为30 nm 的二维层状Fe 纳米片,并研究了其对MgH2的影响。MgH2+5%(质量分数)Fe 的纳米复合材料具有最佳的吸氢和解吸性能。脱氢温度为182.1 ℃,吸氢温度为75 ℃。此外,在200 ℃下,10 分钟内可吸收6%(质量分数)H2。经过几次循环测试,氢含量保持在5%(质量分数)左右。进一步分析表明,Fe 纳米片的加入可以削弱Mg—H 键从而提高MgH2的脱氢性能(图24)。
综上所述,将具有优异催化效果的过渡金属单质引入MgH2中可以有效改善吸脱氢性能,削弱Mg—H 键,降低储氢材料的活化能。但是单一过渡金属的引入,无法实现多元素的协同催化效果,另外过渡金属单质硬度较高,在一定程度上降低了球磨效率,限制了它的应用。
2.3.2 过渡金属氧化物改性
与金属单质相比,金属氧化物具有更高的脆性,通过球磨技术使其更容易分散在MgH2基体中,引入过渡金属氧化物同样可以有效地催化MgH2的吸放氢反应[122-123]。过渡金属氧化物根据所含元素可分为二元、三元两大类。
对于二元过渡金属氧化物,早在1991 年Terzieva 等[124]就报道了Fe2O3和MnO2能够提升储氢材料的吸放氢动力学性能。研究发现,金属氧化物部分还原为金属颗粒,这些金属可加快氢分子的分解,从而促进体系的动力学性能。随后,2001 年Oelerich 等[125]通过简单球磨的方法将各种一元过渡金属氧化物(Sc2O3、TiO2、V2O5、Cr2O3、Mn2O3、Fe3O4、CuO/Al2O3和SiO2)引 入MgH2中,并探讨了它们对Mg基储氢材料吸脱氢性能的影响。在这些氧化物中,添加CuO的MgH2复合材料可吸氢5.5%(质量分数)H2,在300 ℃真空条件下10 min之内实现完全脱氢,添加V2O5和Fe3O4的氢化镁在5 min 之内就可完成放氢。高价态Nb2O5和ZrO2对MgH2体系的吸放氢性能也有显著提升。
然而,由于简单球磨法制备的TiO2纳米片在循环过程中容易发生团聚,严重减少催化活性位点[126]。为了解决这一问题,邹建新团队[127]首次尝试利用纳米尺寸效应和催化剂添加效应,通过溶剂热法设计了一种新型的自组装MgH2/TiO2异质结构纳米复合材料。该复合材料的催化机理如图25 所示:①高比表面积的TiO2纳米片为氢的扩散提供了更多的通道,并为MgH2/Mg提供了大量成核位点。②纳米MgH2均匀负载在TiO2纳米片上,显著抑制了纳米MgH2在吸氢/脱氢过程中的生长和团聚,确保了良好的循环稳定性。③大量氧空位显著提高了TiO2的导电性,并为电子和氢的运输提供了额外的活性位点,进一步改善吸脱氢性能。④多价Ti基催化剂组成的多相界面为MgH2/Mg 提供更多的氢扩散途径和成核位点。性能方面:MgH2/TiO2的起始脱氢温度降至180 ℃。在300 ℃时,MgH2/TiO2的脱氢速率为2.116%(质量分数)/min,是相同条件下纯MgH2的35倍。
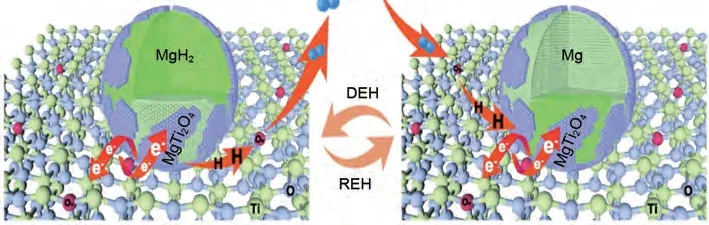
图25 MgH2/TiO2异质结构吸氢和脱氢机理示意图[127]Fig.25 Schematic diagram of the hydrogenation and dehydrogenation mechanisms of MgH2/TiO2 heterostructure[127]
针对三元过渡金属氧化物,第一种是以MgH2-TM(O)/TMO(TM为过渡金属)形式复合。
朱云峰团队等[128]首次通过层状双金属氢氧化物(LDH)前驱体在氢气和氩气氛围下煅烧分别制备了层状Ni/Al2O3和NiO/Al2O3。对比发现Ni/Al2O3对MgH2的吸氢性能具有更加优异的催化作用。如图26 所示,TPD 曲线[图26(a)]表明,MgH2-Ni/Al2O3的起始脱氢温度(190 ℃)远低于MgH2-NiO/Al2O3(240 ℃)和球磨后的MgH2(298 ℃),说明Ni/Al2O3对脱氢的催化活性优于NiO/Al2O3。等温脱氢动力学中也存在类似现象。在275 ℃时,MgH2-5%(质量分数)Ni/Al2O3脱氢动力学更快,在3500 s内脱附5.6%(质量分数)H2,MgH2-5%(质量分数)Ni/Al2O3脱附4.6%(质量分数)H2,球磨后的MgH2几乎无法脱氢[图26(b)]。在250 ℃和300 ℃下[图26(c)、(d)],MgH2-Ni/Al2O 比MgH2-NiO/Al2O3展现出更好的脱氢动力学和更高的储氢量。通过拟合不同的脱氢动力学模型发现[图26(e)],斜率最接近于1的动力学模型为F1模型(一级动力学),即脱氢速控步为氢的重组[129]。通过Arrhenius方程计算的MgH2-5%(质量分数)Ni/Al2O3的脱氢活化能Ea(72.6 kJ/mol)[图26(f)]远低于MgH2(160 kJ/mol)。最后,结合实验和DFT计算证明,该复合材料氢解吸动力学增强主要是由于Ni/Al2O3作为电子受体捕获Mg—H键上的电子,造成MgH2的失稳效应,导致Mg—H键强度减弱,从而改善储氢性能。
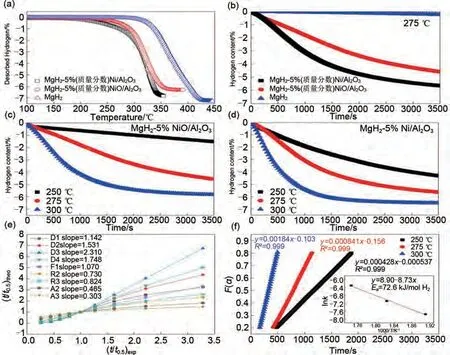
图26 (a) MgH2、MgH2-5%(质量分数)Ni/Al2O3和MgH2-5%(质量分数)NiO/Al2O3的程序升温脱附曲线以及(b) 275 ℃下的等温脱氢曲线;(c) MgH2-5%(质量分数)NiO/Al2O3和(d) MgH2-5%(质量分数)Ni/Al2O3在250 ℃、275 ℃和300 ℃下的等温脱氢曲线;(e) MgH2-5%(质量分数)Ni/Al2O3对应的固相反应机理模型和速率控制步骤;(f) 随时间变化的动力学模型以及由Arrhenius方程得到的随温度变化的速率常数图[128]Fig.26 (a)Temperature-programmed-desorption and (b) isothermal dehydrogenation curves at 275 ℃ of MgH2,MgH2-5%Ni/Al2O3 and MgH2-5%NiO/Al2O3 ; isothermal dehydrogenation curves of (c)MgH2-5%NiO/Al2O3 and (d)MgH2-5%Ni/Al2O3 at different temperatures of 250 ℃, 275 ℃ and 300 ℃; (e)the corresponding solid-state reaction mechanism model and rate-controlling step of MgH2-5%Ni/Al2O3; (f)time dependence of the kinetic modeling equation F(α)and the plot for the temperature-dependent rate constant k, obtained by the Arrhenius equation[128]
武英团队[130]将TiO2引入Sc2O3中,采用溶胶-凝胶法和煅烧法合成了Sc2O3/TiO2催化剂,并将其加入MgH2中以改善MgH2的脱氢动力学性能。计算得到MgH2-5%(质量分数)Sc2O3/TiO2复合材料的脱氢活化能为77.8 kJ/mol,比商用MgH2(160 kJ/mol)低一半。此外,当在MgH2中加入5%(质量分数)的Sc2O3/TiO2催化剂后,复合材料储氢性能显著提高,在300 ℃下1000 s内释放6.21%(质量分数)H2,并且可以在200 ℃下3000 s 内吸收6.08%(质量分数)H2,性能远超Mg/MgH2。优异的储氢性能可归因于均匀分布在Mg颗粒表面的Sc和Ti的协同催化效应和对Mg/MgH2团聚粗化的抑制效应(见图27)。
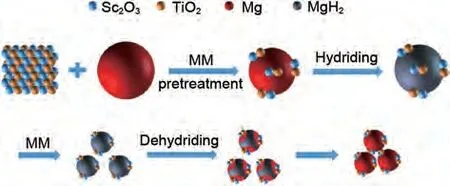
图27 MgH2@5%(质量分数)Sc2O3/TiO2复合材料在吸氢--脱氢过程中的微观结构演变示意图[130]Fig.27 Schematic diagram of the microstructural evolution for the MgH2@5%Sc2O3/TiO2 composite during the hydrogenation-dehydrogenation process [130]
第二种是以MgH2-TMTMO形式复合。
Sazelee等[131]通过高温固相法合成了BaFe12O19,结果表明,MgH2+10%(质量分数)BaFe12O19样品在约270 ℃开始脱氢,比研磨后的MgH2低约70 ℃。MgH2+10%(质量分数)BaFe12O19复合材料能够在150 ℃下10 分钟内吸附4.3%(质量分数)H2,而研磨后的MgH2仅能吸附3.5%(质量分数)H2。此外,掺杂BaFe12O19的MgH2材料的脱氢活化能为115 kJ/mol, 低 于 研 磨 的MgH2(141 kJ/mol)。Zhang 等[132]结合共沉淀法和水热法合成了LiCoO2纳米片,发现它对MgH2的储氢性能具有显著的催化作用。MgH2-LiCoO2体系的初始脱氢温度降至180 ℃,250 ℃下60 min内脱氢量高达5.5%(质量分数)。脱氢活化能降低至(48.5±0.4) kJ/mol,相比MgH2(81.4±5.6 kJ/mol)降低了40.4%。分析表明,LiCoO2在MgH2中均匀分布,并在循环过程中自组装成大量Mg2Co-Mg2CoH5“纳米氢泵”,从而加速了氢的扩散速率,提高了储氢性能(见图28)。
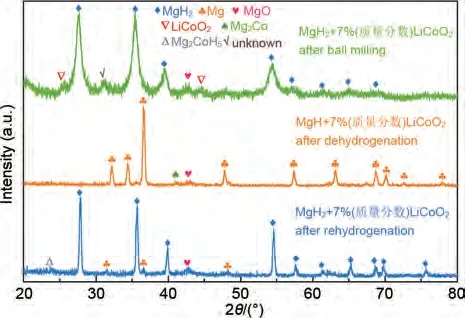
图28 MgH2+7% LiCoO2复合材料球磨后、脱氢后和再吸氢后的XRD谱图[131]Fig.28 XRD patterns of MgH2+7% LiCoO2 composite:after ball milling, after dehydrogenation and after rehydrogenation [131]
鉴于Mg2Co 和Mg2CoH5之间的可逆相变具有的“氢泵”效应可有效改善镁基储氢材料的吸脱氢性能。潘复生团队[133]通过水热和煅烧两步方法成功合成了厚度为60~100 nm 的多层页片状MnCo2O4.5纳米颗粒,以探索二元钴和锰基氧化物对MgH2的催化作用。MgH2-6%(质量分数)MnCo2O4.5复合材料的初始脱氢温度降至285 ℃,在325 ℃下4 min内完全释放6.4%(质量分数)H2。脱氢后的MgH2-6%(质量分数)MnCo2O4.5复合材料在150 ℃下30 min 内吸收4.43%(质量分数)H2,吸脱氢性能相比球磨态MgH2有了显著改善[见图29(a)~(d)],复合材料经30 次吸脱氢循环后吸氢量仅衰减了0.75%(质量分数),脱氢量基本维持在6.24%(质量分数)左右,展现出了优异的循环储氢性能[见图29(e)、(f)]。一些常见的MgH2-三元过渡金属氧化物复合材料的储氢性能如表5所示。
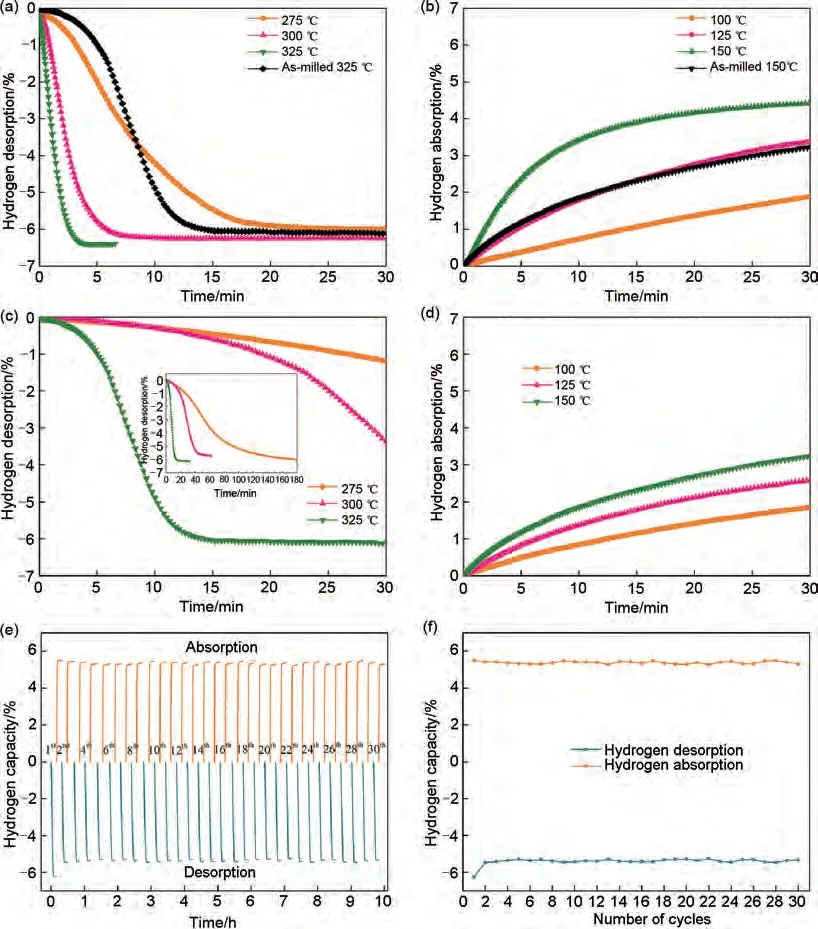
图29 (a) MgH2-6%(质量分数)MnCo2O4.5和(c)碾磨态MgH2的等温脱氢曲线;(b)MgH2-6%(质量分数)MnCo2O4.5和(d)碾磨态MgH2的等温吸氢曲线;(e)MgH2-6%(质量分数)MnCo2O4.5在325 ℃时的脱氢循环曲线;(f)储氢容量的衰变曲线[120]Fig.29 (a)Isothermal dehydrogenation curves of MgH2-6 wt% MnCo2O4.5 and (c)as-milled MgH2; (b)isothermal hydrogenation curves of MgH2-6 wt% MnCo2O4.5 and (d)as-milled MgH2; (e)de/hydrogenation cycle curves of MgH2-6 wt% MnCo2O4.5 at 325 ℃, and (f)decay curves of hydrogen storage capacity[120]
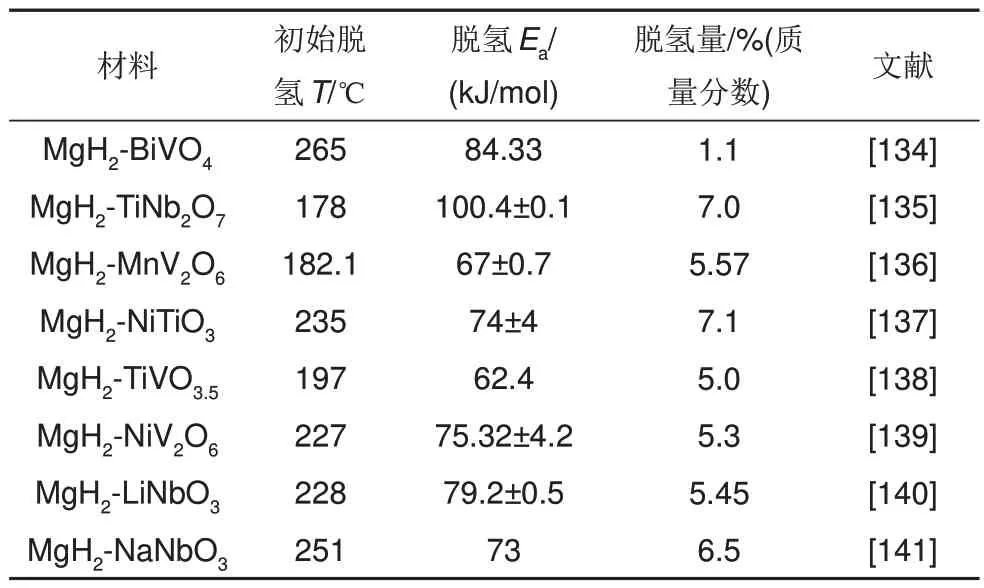
表5 一些MgH2-三元过渡金属氧化物的储氢性能Table 5 Hydrogen storage properties of some MgH2-ternary transition metal oxides
综上所述,多元过渡金属氧化物可以展现出比单一过渡金属更加优异的催化效果,氧空位的引入可以大大加快电子转移,进一步改善储氢材料的热力学/动力学性能。
2.3.3 其他金属化合物
除金属氧化物外,氟化物、硫化物和金属-有机骨架化合物(MOFs)等其他金属化合物由于其特殊的物理和化学性质而表现出显著的催化活性,其对Mg/MgH2体系的催化作用也受到广泛关注。
针对氟化物,Lin 等[142]研究了球磨法制备的不同价态氟化铈掺杂MgH2的氢解吸性能。研究发现,当掺杂2%(摩尔分数)CeF4后,MgH2的氢解吸温度和表观活化能显著降低,这可能是由于CeF4/MgH2界面上形成的新相Ce-F-Mg 和高价Ce 阳离子促进了电子转移所导致的。此外,MgH2-CeF4的解吸活化能从160 kJ/mol降至110 kJ/mol。Mao等[143]采用电弧等离子体法制备核壳结构Mg-MFx(M=V、Ni、La、Ce)纳米复合材料。其中,Mg-NiF2复合材料在相对较低的温度下表现出最佳的吸氢性能,在100 ℃下2 h 内吸氢量达到3.26%(质量分数),在473 K 下60 s 内吸氢量达到3.85%(质量分数),优异的吸氢性能主要是由于电弧蒸发和冷凝过程中Mg粒子上形成了Mg2Ni和MgF2。Mg-VF3复合材料具有最低的初始脱氢温度(674.2 K),MgF2和金属氧化物覆盖在Mg 颗粒上的核壳结构和较少的团聚倾向是储氢性能改善的主要原因。
与单阳离子氟化物相比,由于过渡金属与钾元素的协同催化作用,双阳离子氟化物也被广泛用作镁基储氢材料的优良催化剂。Sulaiman 等[144]将K2NiF6添加到MgH2中以改善其脱氢特性。结果发现,MgH2-5%(质量分数)K2NiF6材料在260 ℃下可以脱氢,KH、KF 和Mg2Ni 催化相的出现共同改善了MgH2的储氢行为(图30)。Yan等[145]首次研究了新型双阳离子金属氟化物K2TaF7对MgH2储氢特性的催化影响。在只有1%(质量分数)K2TaF7掺杂剂的情况下,MgH2的初始脱氢温度降低了约130 ℃,总脱氢量超过7.3%(质量分数)。MgH2-1%(质量分数)K2TaF7复合材料的解吸活化能降低至(107.2±1.2) kJ/mol。在190 ℃,MgH2-1%(质量分数)K2TaF7样品吸氢量达到6.56%(质量分数),而MgH2吸氢量只有3.45%(质量分数)。研究表明,K2TaF7在脱氢过程中可以与MgH2反应,产生共生氢化物KMgH3和TaH0.8,在氢的释放和吸收过程中发挥氢泵的作用(图31)。
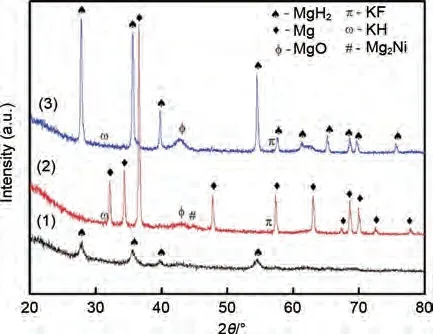
图30 MgH2+5%(质量分数)K2NiF6球磨1 h、450 ℃脱氢和320 ℃吸氢后的XRD谱图[144]Fig.30 XRD patterns of MgH2+5%K2NiF6 composite:after ball milling, after dehydrogenation and after rehydrogenation [144]
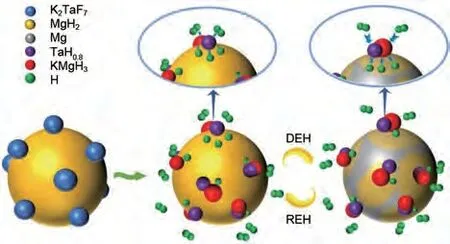
图31 K2TaF7对MgH2的催化机理图[145]Fig.31 Schematic illustration of the catalytic mechanism of the K2TaF7-catalyzed MgH2 composite[145]
针对硫化物,Wang等[146]通过球磨将过渡金属硫化物(TiS2、NbS2、MoS2、MnS、CoS2、CuS)作为催化剂来改善MgH2的储氢性能。结果表明,这些硫化物均能显著提高MgH2的氢解吸动力学。MgH2-TiS2的吸氢和脱氢动力学最好,MgH2-TiS2的初始脱氢温度约为204 ℃,比MgH2低约126 ℃。脱氢活化能降低至50.8 kJ/mol。硫化物的有益催化作用可归因于原位形成的MgS、TiH2、NbH、Mo、Mn、Mg2CoH5和MgCu2相。
基于新型双阳离子金属硫化物,韩树民团队[147]设计了一种中空球囊结构的三元过渡金属硫化物FeNi2S4作为MgH2的催化剂。值得注意的是,MgH2-FeNi2S4复合材料中活性物质Mg2Ni/Mg2NiH4、MgS 和Fe 的协同催化作用显著提高了MgH2的脱氢/加氢性能(图32)。MgH2-FeNi2S4复合材料在373 K下1 h内的吸氢量达到4.02%(质量分数),与球磨态MgH2(0.67%,质量分数)形成鲜明对比。脱氢过程中,MgH2-FeNi2S4复合材料的初始脱氢温度比球磨态MgH2低80 K,脱氢活化能比MgH2(161.2 kJ/mol)降低了95.7 kJ/mol。
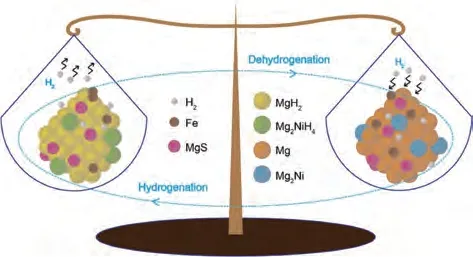
图32 MgH2-FeNi2S4复合材料的加氢/脱氢过程示意图 [147]Fig.32 Schematic diagram of the hydrogenation/dehydrogenation processes of the MgH2-FeNi2S4 composite [147]
除了氟化物与硫化物,金属-有机骨架化合物在改善镁基储氢材料性能方面也得到了深入的研究。金属-有机骨架(MOFs)化合物是以金属离子为连接中心,有机配体为桥联体,通过配位键、氢键、π-π键、范德华力等作用力,自组装而成的具有周期性的多孔配位聚合物[148]。由于具有充足的金属中心、较大的比表面积、丰富的孔道结构、可设计的拓扑单元以及灵活可调的组成成分,MOFs的应用研究延伸至众多领域[149-151]。2003 年,美国密歇根大学的Yaghi 教授等[152]首次报道了MOF-5 的储氢性能,MOF-5 是由Zn2+盐和1, 4-对苯二甲酸有机配体合成的多孔八面体结构骨架,经过测试发现在77 K、1 bar(1 bar=100 kPa)外界条件下,MOF-5 的储氢量为1.3%(质量分数)。但正如引言中所提到的,MOF 材料只能在极低温度下实现储氢,这与国际能源机构规定的在室温下实现高效储氢的要求相去甚远,但鉴于其比表面积大、孔隙率高、活性金属位点多等优点,近年来作为催化剂、催化剂载体被广泛应用于改善镁基储氢材料储氢性能的研究当中[153]。
MOF材料的中心原子大多为过渡金属(Co、Ni等)或具有催化效果的非过渡金属(Mg、Zn 等),因此其自身即可作为催化剂用于改善Mg的储氢性能。
MOF 材料ZIF-8、ZIF-67 和MOF-74 的中心原子分别为Zn、Co和Mg,Wang等[154]采用沉积还原法制备了Mg/MOF(MOF=ZIF-8、ZIF-67、MOF-74)储氢复合材料,如图33(a)所示,在175 ℃下Mg/ZIF-67、Mg/ZIF-8 和Mg/MOF-74 分别在2051 s、3759 s和3966 s内吸收2%(质量分数)H2,分别比未添加MOF 材料的Mg 纳米颗粒快3124 s、1708 s 和1915 s,吸氢速度的提升很好地证明了Mg-MOF复合体系的优异催化效果。在300 ℃时研究了材料的脱氢动力学性能[见图33(b)],5000 s内Mg、Mg/MOF-74、Mg/ZIF-8 和Mg/ZIF-67 分别释放了0.6%(质量分数)、1.2%(质量分数)、2.7%(质量分数)和3.7%(质量分数)H2,Mg/ZIF-67具有最为可观的脱氢量。此外,Mg/ZIF-67复合材料在350 ℃时最大储氢量达到了5.30%(质量分数),即使在100次氢气吸收/解吸循环后,吸脱氢量没有任何衰减。
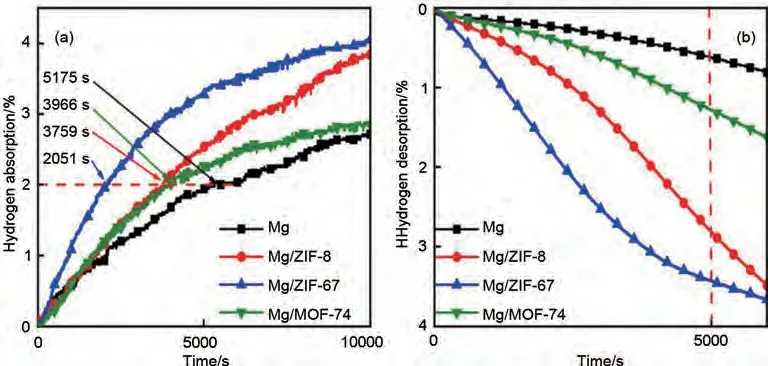
图33 Mg、Mg/ZIF-8、Mg/ZIF-67和Mg/MOF-74复合材料的等温氢化曲线[154]:(a)175 ℃吸氢过程;(b)300 ℃脱氢过程Fig.33 Isothermal hydriding curves of the Mg, Mg/ZIF-8, Mg/ZIF-67, and Mg/MOF-74composites [154]: (a)hydrogenation process at 175 ℃; (b)dehydrogenation process at 300 ℃
以过渡金属作为中心原子制备出的MOF 可以大大增加比表面积,产生更多的吸脱氢扩散通道,从而大大改善储氢性能。朱云峰团队等[155]通过简单的水热反应首次制备了结构稳定的新型花状Ni-MOF材料,并将其引入MgH2中,研究表明该材料在提高MgH2的储氢性能方面表现出优异的催化活性。MgH2-5%(质量分数)Ni-MOF 的峰值脱氢温度比纯MgH2低78 ℃。该复合材料在300 ℃下600 s内释放6.4%(质量分数)H2,并在150 ℃吸附约5.7%(质量分数)H2。花状Ni-MOF 的高催化活性可归因于原位生成的Mg2Ni/Mg2NiH4、MgO 纳米颗粒、无定形C和剩余的层状Ni-MOF的结合作用(见图34)。
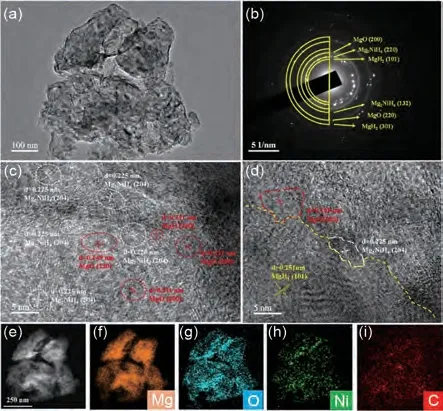
图34 (a) 明场TEM图; (b)SAED图; (c)、(d) HRTEM图像; (e)~(i) 暗场图和11次氢化后MgH2-5%(质量分数)Ni-MOF的EDS图[155]Fig.34 (a)Typical bright field TEM micrograph; (b)the corresponding SAED pattern; (c)、(d)the HRTEM images, (e)—(i)the dark field micrographs and the corresponding elemental mappings of MgH2-5% Ni-MOF after 11th hydrogenation [155]
此外,具有较大比表面积和丰富多孔结构的MOF 材料具有大量的新鲜表面,可作为过渡金属和Mg的高效载体,通过过渡金属与MOF材料的协同作用改善镁基材料的储氢性能。
郭进团队[156]以ZIF-67 为催化剂载体,采用Mg和过渡金属Nb 共沉积还原的方法成功制备了MgNb/ZIF-67 复合材料,并研究了Nb 和ZIF-67 对Mg 储氢性能的协同作用。如图35(a)所示,在175 ℃下,MgNb 和MgNb/ZIF-67 吸附2%(质量分数)H2分别需要23 min 和10 min,MgNb/ZIF-67 具有更快的吸氢速率,在275 ℃的脱氢过程中[图35(b)],MgNb/ZIF-67仅需要8 min就可释放出3%(质量分数)H2,而MgNb 需要23 min 才能达到相同的脱氢量。MgNb/ZIF-67更为迅速的吸脱氢速率证实了过渡金属与MOF 材料之间存在协同催化作用。但是制备出的Mg颗粒存在严重的团聚现象[见图36(a)],而MgNb/ZIF-67 具有丰富的多孔结构且无明显团聚现象[见图36(b)],由100次循环后复合材料的TEM图[图36(c)]可看出,ZIF-67中心原子Co所延伸的晶面负载了大量的晶格Mg,剩余的少量Mg、Nb 和部分氧化的NbO 紧密接触,形成了CoMg2-Mg(Nb)核壳结构,该结构具有纳米限域的效果,保证了MgNb/ZIF-67稳定的循环储氢性能。
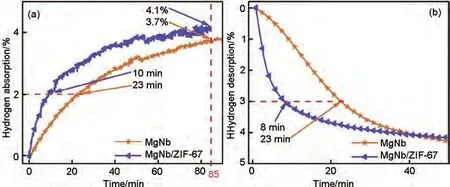
图35 MgNb和MgNb/ZIF-67纳米复合材料在(a)175 ℃和(b)275 ℃下的等温吸/脱氢曲线[156]Fig.35 Isothermal hydrogenation/dehydrogenation curves of MgNb and MgNb/ZIF-67 nanocomposites at (a)175 ℃ and (b)275 ℃ [156]

图36 (a)、(b) Mg和MgNb/ZIF-67纳米复合材料的SEM图; (c)~(e)MgNb/ZIF-67 100次吸/脱氢循环后的TEM图像和SAED图[156]Fig.36 (a)、(b) SEM micrographs of the Mg and MgNb/ZIF-67 nanocomposites (c)—(e)TEM images and SAED of MgNb/ZIF-67 after 100 hydrogen absorption/desorption cycles [156]
Zhang 等[157]通过水热反应和随后的煅烧过程,成功将Nb2O5纳米粒子负载在了菱形十二面体金属有机骨架上,该纳米粒子的平均粒径为10 nm(图37)。如图37 所示,复合了7%(质量分数)Nb2O5@MOF 的MgH2在181.9 ℃开始脱氢,在275 ℃和250 ℃时分别在2.6 min和6.3 min内释放出6.2%(质量分数)H2。此外,Nb2O5颗粒均匀分布在MgH2基体表面,并与MOF通过协同催化作用提高了MgH2的储氢性能。
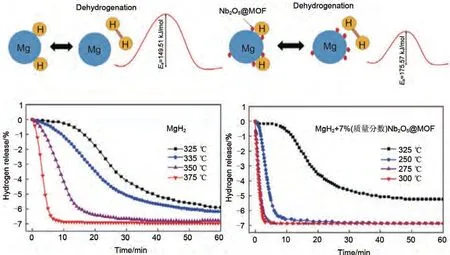
图37 Nb2O5和MOF协同改善MgH2的脱氢性能机理图和脱氢曲线[157]Fig.37 The mechanism diagram and corresponding dehydrogenation curve of MgH2 improved by Nb2O5 and MOF [157]
总体来说,MOF 自身可作为催化剂,也可作为其他催化剂或者Mg/MgH2的成核位点用于改善储氢材料的动力学/热力学性能。此外,相比过渡金属单质的简单添加,以过渡金属为中心原子的MOF 材料具有更为优异的结构,吸脱氢性能也有了进一步改善。部分MOF改性的MgH2的储氢性能汇总见表6。
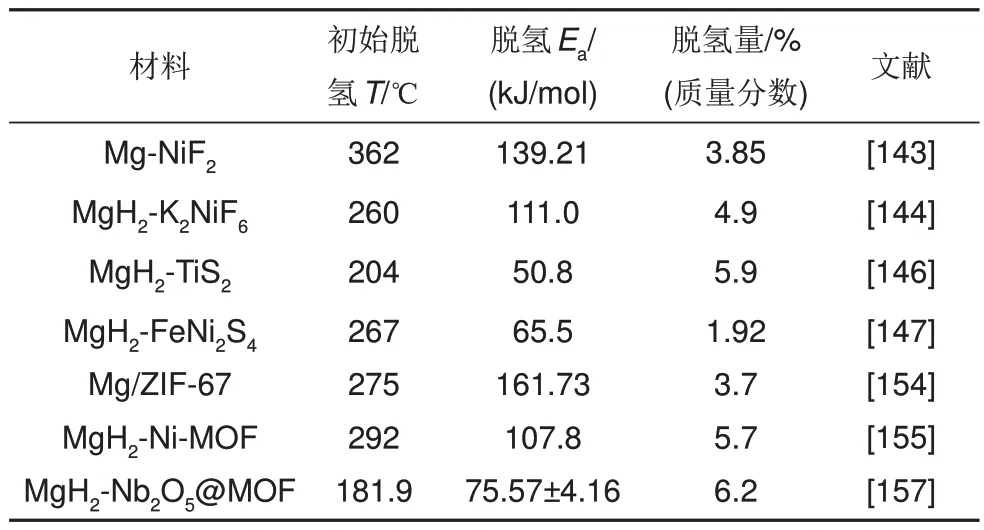
表6 部分其他金属化合物改性的MgH2的储氢性能汇总表Table 6 Summary of hydrogen storage properties of MgH2 modified by some other metal compounds
2.4 金属与碳基复合催化剂改性
催化剂的效果不仅与其本身性质有关,其形态、粒径、分散性等因素也显著影响其催化活性。碳基材料是一种良好的催化剂载体,更重要的是碳基材料可以有效抑制MgH2颗粒的团聚和长大。碳基材料(碳纳米管、石墨烯、MXene等)具有良好的导电性、化学稳定性,较高的比表面积等一系列优点,广泛应用于催化和储氢领域。与其他储氢材料相比,碳基材料具有较小的质量密度,有利于提升整个体系的质量储氢密度,因此受到了研究者越来越多的关注[158-159]。
2.4.1 碳纳米管
碳纳米管(carbon nanotube, CNT)是碳材料家族的一位新成员,系Iijima[160]于1991 年首次发现。CNT是将单层石墨烯卷曲闭合,呈六边形排列的碳原子构成圆管,单壁碳纳米管(SWCNT)只有一层,多壁碳纳米管(MWCNT)为数层的同轴层状中空结构圆管,具有重量轻、化学稳定性高、机械强度高等特点,CNT层间可包覆MgH2/Mg防止团聚现象,而且纳米材料所具有的量子效应、表面效应等特性使其对氢气吸附具有增强效应。
继Iijima 首次发现CNT 后,研究人员对CNT在储氢方面的应用进行了各种探索。Kajiura 等[161]发现SWCNT、MWCNT 在环境温度和高达8 MPa的压力下储氢量没有超过0.43%(质量分数);Ritschel等[162]对不同碳纳米结构的储氢能力进行研究发现,纯化的SWCNT在室温和4.5 MPa的压力下可逆储氢容量仅为0.63%(质量分数),可以看出碳纳米管需要在低温高压下才能具有一定的吸氢能力。Mosquera-Vargas 等[163]研究发现,具有729.4 m2/g 比表面积的纯化MWCNT 在室温和12.79 kPa低压条件下最大可以吸收3.46%(质量分数)H2,但储氢量仍不理想。近年来随着MgH2高容量储氢材料的快速发展,许多研究人员开始将拥有丰富成核位点的碳纳米管作为金属的优良载体,用于改善MgH2的吸脱氢性能,取得了一系列成果。
Liu 等[164]将纳米级双向催化剂Co/Pd 均匀地分散在少壁的竹节状碳纳米管(B-CNTs)上,并通过球磨引入MgH2中。该复合材料对氢的吸附和解吸均表现出较好的催化效果。提出了如下的储氢机理(图38):①在球磨过程中,B-CNTs被机械力撕裂成少壁碳片。这些支撑Co/PdNPs的撕裂碳片覆盖在MgH2颗粒表面,形成区隔层,防止了MgH2纳米颗粒的团聚和烧结,提高了循环稳定性。②B-CNTs具有大直径(>100 nm)和高比表面积(1468 m2/g),促进了Co/PdNPs 的均匀分散,增加了自身与MgH2纳米颗粒的接触面积,为氢气扩散提供了新的通道。③在氢化过程中,元素Pd起着主要的加速作用。氢原子在Pd/Mg界面优先扩散。而在脱氢过程中,Mg2Co与Mg2CoH5以及Mg Pd合金之间的相变降低了扩散势垒从而促进氢原子释放。MgH2-Co/Pd@B-CNTs 复合材料在198.9 ℃时开始脱氢,比研磨后的MgH2低132.4 ℃。在Co/Pd@B-CNTs的催化作用下,MgH2的脱氢活化能由178.0 kJ/mol降低到76.66 kJ/mol。同时该复合材料也表现出了优异的动力学性能,在250 ℃下10 s 内快速吸附6.68%(质量分数)H2,即使在50 ℃下100 s 内也有1.91%(质量分数)吸氢量,实现了吸脱氢量和热/动力学性能的双重调控。
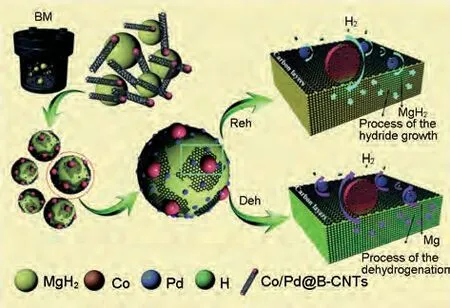
图38 Co/Pd@B-CNTs对MgH2脱氢和吸氢的“双向催化”机理示意图[164]Fig.38 Schematic illustration for the “bidirectional catalysis” mechanism of Co/Pd@B-CNTs on the dehydrogenation and hydrogenation of MgH2[164]
Zhang 等[165]采用简单的凝胶过滤和煅烧法制备了Ni@CNT 和多孔Ni3ZnC0.7颗粒的复合催化剂(Ni3ZnC0.7/Ni@CNT),并通过高效球磨引入高纯MgH2中。如图39 所示,MgH2-5%(质量分数)Ni3ZnC0.7/Ni@CNT复合材料经过第一次氢解吸后,会原位分解为Mg2Ni 和Zn。Zn/MgZn2的可逆相变以及Mg2Ni/Mg2NiH4的“氢泵”效应能够在氢吸附和解吸过程中为氢提供更多的成核位点和扩散通道。Ni@CNT的存在也可以有效抑制纳米复合材料在吸脱氢循环过程中的团聚和烧结。该材料在80 ℃下60 min内能吸附2.34%(质量分数)H2,在300 ℃时释放约5.36%(质量分数)H2。MgH2-5%(质量分数)Ni3ZnC0.7/Ni@CNT复合材料的吸氢活化能和脱氢活化能分别降至37.28 kJ/mol 和84.22 kJ/mol[图40(a)、(b)]。此外,MgH2-5%(质量分数)Ni3ZnC0.7/Ni@CNT复合材料的脱氢起始温度降至约110 ℃,而纯MgH2在约400 ℃高温下才能开始脱氢[图40(c)]。
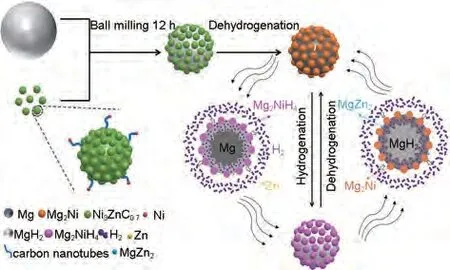
图39 MgH2-5%(质量分数)Ni3ZnC0.7/Ni@CNT纳米复合材料吸/脱氢过程中催化剂催化机理示意图[165]Fig.39 Schematic diagram of the catalytic mechanism of catalysts during the hydrogenation/dehydrogenation processes of the MgH2-5 wt%Ni3ZnC0.7/Ni@CNT nanocomposite[165]
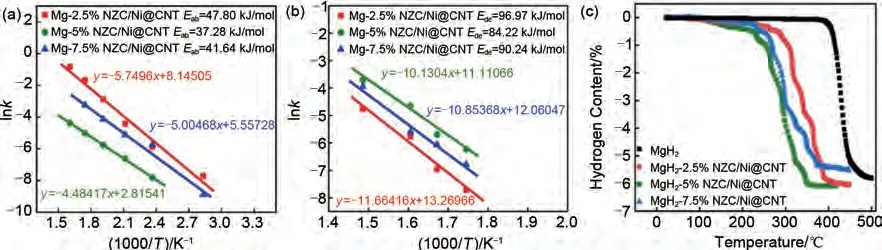
图40 氢化态(a)和脱氢态(b) MgH2-xNZC/Ni@CNT(x=2.5%,5%,7.5%,质量分数)复合材料的相应lnk与1000/T图;(c) MgH2-xNZC/Ni@CNT(x=2.5%,5%,7.5%,质量分数)和商用MgH2的TPD曲线[165]Fig.40 The corresponding lnk vs 1000/T plots for hydrogenated (a) and dehydrogenated (b) MgH2-xNZC/Ni@CNT (x=2.5%, 5%, 7.5%)composites.Comparison of TPD curves (c) of MgH2-xNZC/Ni@CNT (x=2.5%, 5%,7.5%)and MgH2[165]
可见,碳纳米管可制备成比表面积较大的竹节状等形貌从而作为过渡金属催化剂的高效载体用于改善镁基储氢材料的储氢性能。
2.4.2 石墨烯
石墨烯(Graphene)是通过碳原子的sp²杂化紧密堆积所形成的具有二维六元环单层结构的碳材料。自2004年曼彻斯特大学制备出能够在室温下稳定存在的石墨烯薄片材料后,其研究得到了快速的发展。2015年,Zhang等[166]采用实验和第一性原理计算的方法,以晶格组成为Mg4H8的物理模型首次系统研究了Graphene 对MgH2脱氢性能的催化作用和机理。研究发现Graphene纳米片分散包覆在MgH2颗粒外表面,有效抑制了球磨过程中MgH2颗粒的团聚。石墨烯改性的机理在于同时降低了MgH2的脱氢焓和脱氢活化能,活化能从169.25 kJ/mol 降至90.01 kJ/mol(图41)。
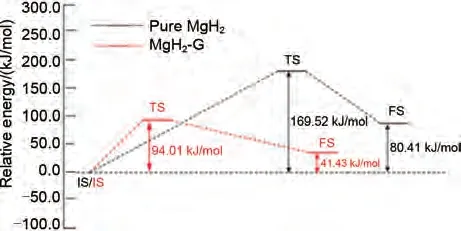
图41 Mg4H8放氢过程的焓变 (a) 和石墨烯引入MgH2的活化能 (b)[166]Fig.41 (a) enthalpy change of graphene-doped Mg4H8 dehydrogenation and (b) activation energy of MgH2[166]
与碳纳米管类似,石墨烯也具有较高的比表面积,可以为其他过渡金属等催化剂提供丰富的负载位点,从而协同改善MgH2的吸脱氢性能。陈萍团队[167]通过真空煅烧和机械球磨的方法成功制备了MgH2+10%(质量分数)Fe-Ni@3DG(三维石墨烯)复合材料。如图42(a)、(b)所示,MgH2+10%(质量分数)Fe-Ni@3DG 复合体系可以在300 ℃下100 s 内吸收6.35%(质量分数)H2,并在500 s 内释放5.13%(质量分数)H2。此外,在7 次循环吸脱氢性能测试中该体系能够在10分钟内吸收6.5%(质量分数)H2并释放5.7%(质量分数)H2[图42(d)],明显优于球磨态MgH2的循环性能[图42(c)],表现出优异的循环稳定性。两种过渡金属和石墨烯的多重催化作用进一步改善了MgH2的吸脱氢性能。
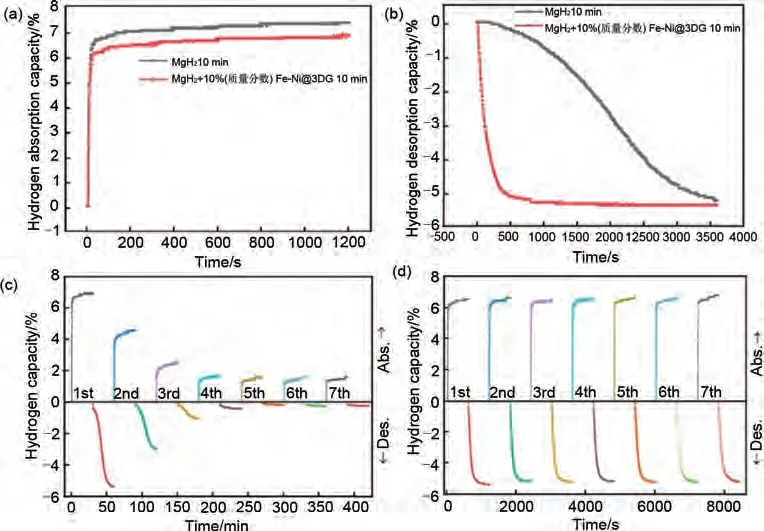
图42 MgH2与MgH2+10%(质量分数)Fe-Ni@3DG吸脱氢曲线:(a) 等温吸氢;(b) 等温脱氢;(c) MgH2循环性能图;(d) MgH2+10%(质量分数)Fe-Ni@3DG循环性能图[167]Fig.42 Adsorption dehydrogenation curves of MgH2 and MgH2+10%Fe-Ni@3DG: (a) isothermal hydrogen absorption; (b) isothermal dehydrogenation; (c) cyclic performance diagram of MgH2; (d) cyclic performance diagram of MgH2+10%Fe-Ni@3DG[167]
因此,石墨烯自身可作为催化剂改善储氢材料的活化能、焓变等参数,也可作为其他催化剂的载体通过协同催化作用改善储氢材料的吸脱氢循环性能。
2.4.3 金属碳化物/氮化物
二维过渡金属碳化物/氮化物(MXene)是一种具有层状结构[114]的新兴金属碳/氮基材料,具有化学耐久性、导热性能好、力学性能好等优点,在提高MgH2储氢性能方面显示出较高的催化活性[168-170]。对于二维材料MXene Ti3C2的研究开始于2011年[171],随后在MXene 的合成、性能和多用途应用等方面的研究开始不断深入。MXene 的通式为MnXTx,其中M为部分过渡金属,如Ti、Mo、Nb、V、Cr、Zr、Ta 等,n=1~4,X 为C 或N,Tx 为表面基团。过渡金属原子与碳原子以交替分层方式排列的结构配置,使MXene具有很高的成分多样性。
然而,与其他二维材料类似,由于表面终端基团(—OH、—O、F)和范德华相互作用引起的层间耦合倾向,MXene 薄片在干燥过程中容易重新堆叠,导致用于锚定MgH2/Mg 材料的自由表面产生巨大损失[172]。此外,MXene 中大量的含氧端(—OH、—O)会与MgH2/Mg 纳米颗粒反应生成MgO或Mg(OH)2,导致负载量和脱氢动力学降低。
为克服以上问题,Zhu等[173]利用具有折叠纳米片形态的分层Ti3C2Tx 作为支撑材料,锚定了超分散的MgH2/Mg 纳米颗粒。通过分散的Ti3C2Tx 纳米片与十六烷基三甲基溴化铵(CTAB)分子之间的静电相互作用制备了3D结构的Ti3C2Tx。随后,通过前驱体MgBu2自下向上自组装的方法在煅烧Ti3C2薄片(Ti-MX)表面原位合成了超分散的MgH2NPs。得到的MgH2@Ti-MX 表现出快速的脱氢动力学,并且具有优异的结构和循环稳定性。由图43(a)~(c)可以看出,含有60%(质量分数)MgH2NPs 的复合材料在140 ℃时开始脱氢,在150 ℃时2.5 h 内能够释放3.0%(质量分数)H2。此外,在200 ℃下循环60 次后仍然保持了高达4.0%(质量分数)H2的可逆储氢容量,动力学方面没有明显损失。纳米束缚和MgH2/Mg与Ti-MX之间的多相界面所引起的纳米尺寸效应,特别是原位形成的催化相TiH2,是该材料优越的吸氢性能的主要原因。
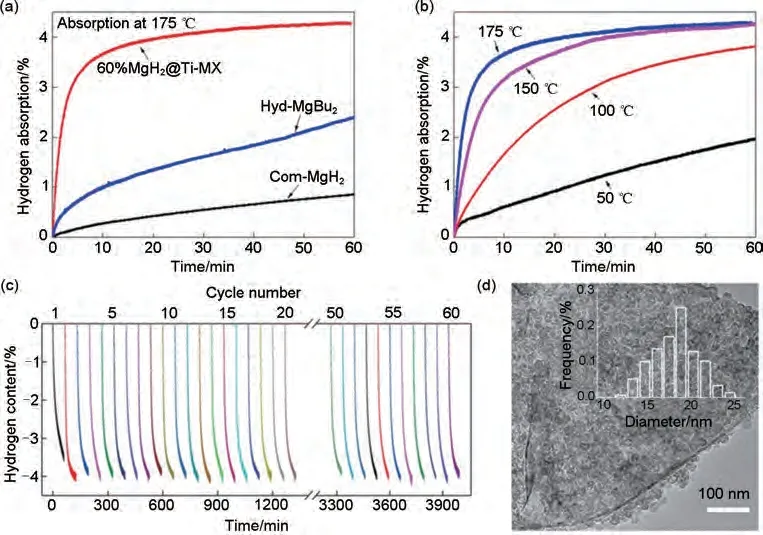
图43 (a)商用MgH2、hyd-MgBu2和60%(质量分数)MgH2@Ti-MX在175 ℃的等温吸氢曲线; (b) 60%(质量分数)MgH2@Ti-MX在不同温度下的等温吸氢曲线; (c) 60%(质量分数)MgH2@Ti-MX在200℃下的循环脱氢曲线;(d) 60%(质量分数)MgH2@Ti-MX在200℃下60次脱氢循环后的典型TEM图像[173]Fig.43 (a) Isothermal hydrogenation curves of commercial MgH2, hyd-MgBu2, and 60% MgH2@Ti-MX at 175 ℃and (b) isothermal hydrogenation curves of 60% MgH2@Ti-MX at different temperatures; (c) cycling dehydrogenation curves of 60% MgH2@Ti-MX at 200 ℃, and (d) typical TEM image of 60% MgH2@Ti-MX after 60 de/hydrogenation cycles at 200 ℃[173]
Liu 等[174]通 过 剥 离 法 制 备 了V2C MXene 与Ti3C2MXene,之后将其与MgH2进行球磨制备了MgH2-2V2C/Ti3C2复合材料。加入10%(质量分数)2V2C/Ti3C2的MgH2在180 ℃左右开始脱氢,在225 ℃下60 min 内可脱附5.1%(质量分数)H2,并且在40 ℃室温条件下20 s 内可吸附5.1%(质量分数)H2。6.3%(质量分数)的可逆储氢量在10 次循环中没有明显下降,表现出了优异的循环稳定性。图44(a)表明在解吸过程中,氢原子或分子优先通过MgH2/V2C/Ti3C2三晶界转移,在吸附过程中,氢原子或分子优先通过Mg/Ti3C2界面转移。图44(b)显示了纯MgH2和MgH2-2V2C/Ti3C2体系分解所需的能量。纯MgH2的脱氢活化能为127 kJ/mol,反应焓为78 kJ/mol,需要跳过49 kJ/mol势垒。对于MgH2-2V2C/Ti3C2体系,脱氢活化能降至79 kJ/mol,反应焓为76 kJ/mol,跳过势垒仅为3 kJ/mol。复合材料较低的活化能以及V2C/Ti3C2对MgH2的轻微失稳效应共同促进了MgH2储氢性能的提高。
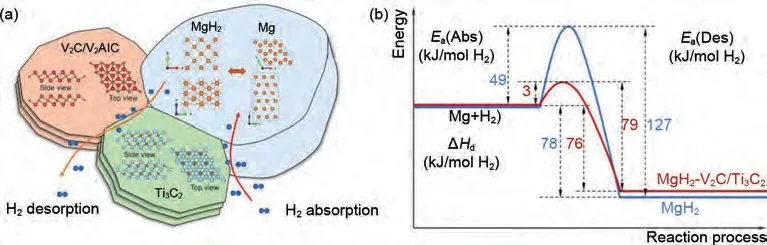
图44 (a) 加入V2C/Ti3C2后MgH2的氢解吸机理示意图;(b) MgH2和MgH2-V2C/Ti3C2的能垒对比示意图[174]Fig.44 (a) Schematic pictures showing the hydrogen desorption and absorption mechanisms of MgH2 with addition of V2C/Ti3C2 and (b) schematic diagram comparatively displaying the energy barriers for as-milled MgH2 and MgH2-V2C/Ti3C2[174]
可见,具有多层结构的MXene材料引入MgH2中可以为氢的吸附和解吸提供丰富的成核位点与扩散通道,将其与一些过渡金属复合,也可以有效改善MgH2的热力学/动力学性能。
综上所述,碳材料可以有效地改善镁基储氢材料的储氢性能,一方面可以为Mg/MgH2提供大量的新鲜表面并对其进行包覆,既减小了颗粒尺寸又起到了抑制团聚的作用;另一方面可以将催化剂负载到碳材料上,既可以减小催化剂的颗粒大小,也可以使催化剂更好地分散到镁基复合材料中。因此,在改善镁基复合材料的储氢性能方面,碳材料起到至关重要的作用。
部分金属与碳基复合催化剂改性的MgH2的储氢性能如表7所示。
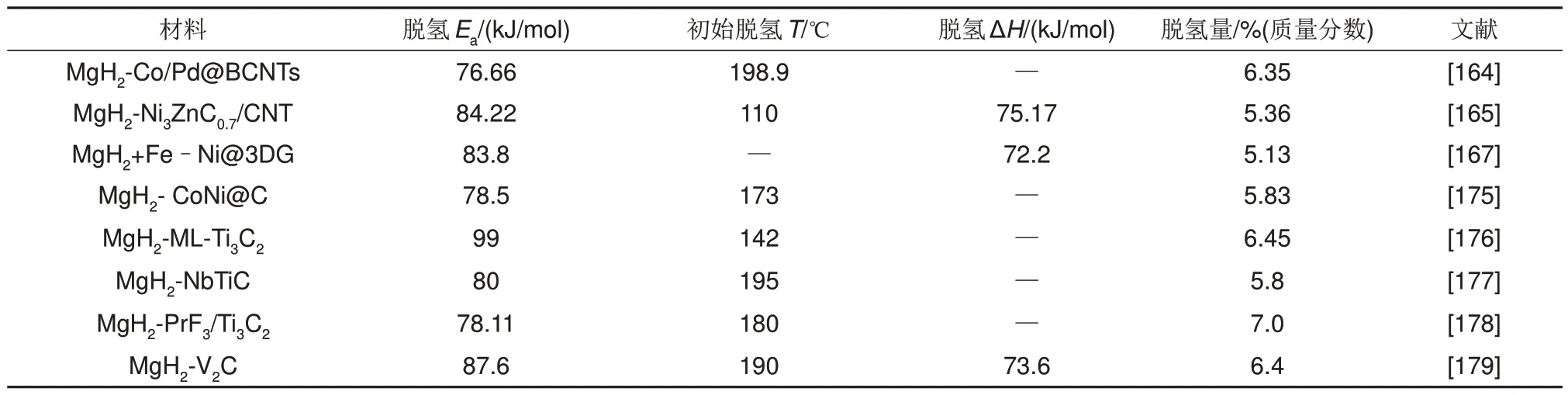
表7 部分金属与碳基复合催化剂改性的MgH2的储氢性能汇总表Table 7 Hydrogen storage performance of MgH2 modified by some metal and carbon based composite catalysts
2.5 高熵合金催化剂改性
合金化改性方面,镁基HEA 受到了广泛的研究,但由于多个金属的引入,合金储氢量发生了明显下降。为了解决此问题,学者们提出了将不含镁的HEA 直接引入MgH2中,由于MgH2本身就是具有可观储氢量的饱和吸氢态,从而使MgH2-HEA复合体系兼顾了理想的吸脱氢量和多个金属元素的多重催化效应。近年来,得到了储氢领域相关科研工作者的深入研究。
潘复生团队[180]采用熔炼- 雾化法制备FeCoNiCrMn高熵合金(HEA),通过球磨引入MgH2中成功制备了MgH2-5%(质量分数)FeCoNiCrMn 复合储氢体系。图45(a)分别显示了MgH2和MgH2-5%(质量分数)FeCoNiCrMn 的TPD曲线。可以清楚地看到,HEA使MgH2的初始脱氢温度(278.2 ℃)降低至209.1 ℃。吸脱氢量方面,图45(b)表明MgH2-5%(质量分数)FeCoNiCrMn 复合材料在250 ℃、300 ℃和350 ℃下脱氢量分别为0.72%、3.76%和5.98%。在相同条件下,研磨的MgH2的脱氢量为0、0.29%和5.21%,在450 ℃时,MgH2-5%(质量分数)FeCoNiCrMn 的脱氢量达到6.1%(质量分数),说明HEA的加入对储氢能力的抑制并不严重。图45(c)显示了MgH2-5%(质量分数)FeCoNiCrMn体系在260 ℃、280 ℃、300 ℃和325 ℃温度下的等温脱氢曲线。可以看出,在260 ℃下30 分钟内脱附了4.8%(质量分数)H2。而研磨态MgH2即使在高达300 ℃条件下也几乎不脱氢[图45(e)]。图45(d)显示了MgH2-5%(质量分数)FeCoNiCrMn 体系在50 ℃、100 ℃、150 ℃和250 ℃温度下的等温吸氢曲线。在250 ℃下22 min 内的吸氢量达到6.0%(质量分数),而在相同温度条件下,MgH2的吸氢量仅有4.0%(质量分数)[见图45(f)]。MgH2-5%(质量分数)FeCoNiCrMn 一系列优异性能证明了MgH2-高熵合金体系相比含镁高熵合金体系更加可观的储氢量。
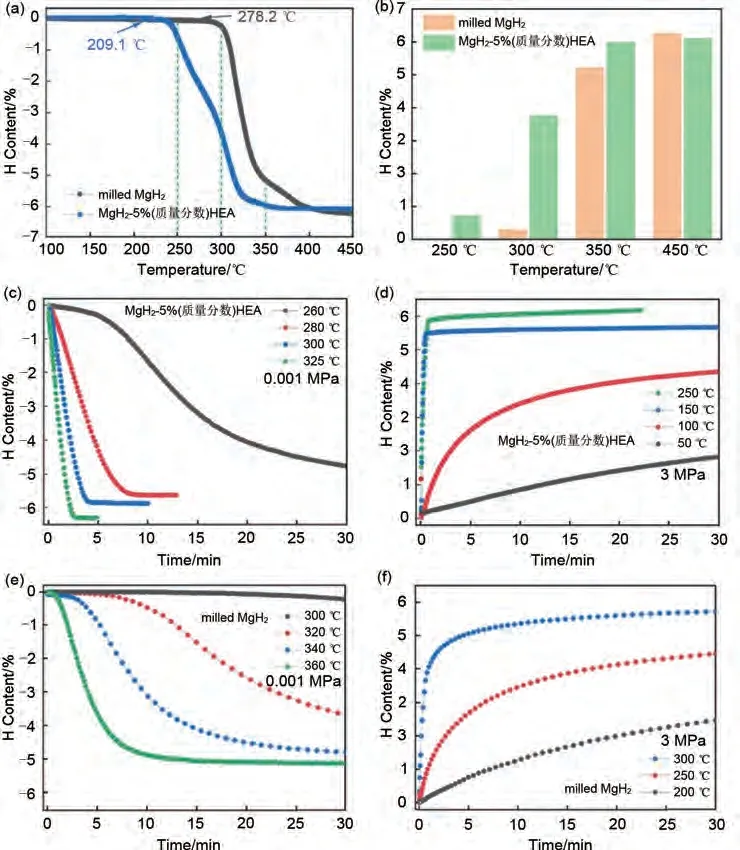
图45 MgH2-5%(质量分数)FeCoNiCrMn和研磨态MgH2的性能比较[180]:(a)TPD曲线;(b)加热到不同温度时的脱氢量;(c)、(e)等温脱氢曲线;(d)、(f)等温吸氢曲线Fig.45 Comparison of MgH2-5%FeCoNiCrMn and milled MgH2[180]: (a) TPD curves; (b) dehydrogenation content when heated to different temperatures; (c)、(e) isothermal dehydrogenation curves ; (d)、(f) isothermal hydrogenation curves
Zhang 等[181]制备了TiVNbZrFe、TiVNbZrNi 和TiVNbCrNi 用于改善 MgH2的储氢性能,TiVNbZrFe 催化效果最佳。MgH2-TiVNbZrFe 体系在大约209 ℃开始释放氢气,比纯MgH2低近170 ℃,在100 次循环内具有高达6.16%(质量分数)的可逆储氢量,脱氢活化能降至63.03 kJ/mol。Verma 等[182]通过氢氧化钠浸泡制备了MgH2-Al20Cr16Mn16Fe16Co16Ni16(LHEA),相比未浸泡MgH2-Al20Cr16Mn16Fe16Co16Ni16(HEA), MgH2-Al20Cr16Mn16Fe16Co16Ni16(LHEA)的初始脱氢温度及最大脱氢量由MgH2-Al20Cr16Mn16Fe16Co16Ni16(HEA)的345 ℃进一步降至338 ℃,最大脱氢量相比HEA 催化体系(6.3%,质量分数)增加至6.8%(质量分数)。Cr、Mn、Fe、Co、Ni的协同催化所产生的“鸡尾酒效应”达到增强催化效果的目的。此外,由于多种元素随机排列,表面非常不均匀,为MgH2脱氢和再氢化过程中氢的扩散提供了更丰富的活性位点。具体催化机理如图46所示。
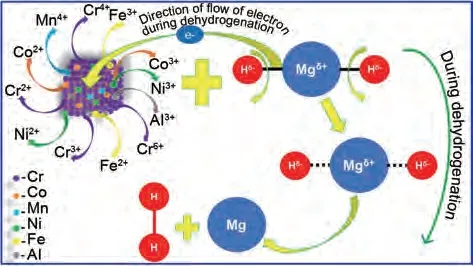
图46 MgH2-LHEA体系的催化机理图[181]Fig.46 Schematic representation of catalytic mechanism of catalyst (LHEA) during dehydrogenation of MgH2[181]
可以看出,MgH2-HEA复合体系相比含镁HEA展现出更为优异的吸脱氢性能,但HEA 组成元素的选择与合金熔炼条件的优化需要进一步深入探索。
3 总结与展望
以青海察尔汗盐湖、茶卡盐湖为代表的卤水盐湖拥有丰富的镁资源,保有储量高达52.29 亿吨,占全国已探明储量的96.7%。因此探索镁元素的高值化利用对盐湖产业高质量发展具有重要意义。
镁基储氢材料储氢容量高,价格低廉,是一类具有极大发展潜力的镁基功能材料。然而,镁基储氢材料的实际应用仍然受到放氢温度较高、动力学较差的限制。学者们采用合金化、纳米化、催化剂掺杂等方法来改善镁基材料的吸放氢热/动力学性能,取得了卓有成效的研究成果。
(1)合金化通过改变镁基材料反应路径,降低吸脱氢活化能和焓来改善动力/热力学性能。然而,目前的镁基合金储氢密度较低,需要进一步提高体系的储氢密度。
(2)纳米化技术在调控MgH2的热/动力学和可逆稳定性方面也起着重要作用。研究表明,兼具“纳米限域”和催化活性的高孔隙率、轻质框架材料是制备纳米Mg 材料的关键影响因素。因此,高稳定性的多孔MOF 及其衍生物是未来制备纳米镁基材料的理想载体。
(3)金属氧化物、氟化物、硫化物等催化剂能够显著提升镁基储氢材料的吸放氢热/动力学性能。添加剂本身或在制备/吸放氢过程中形成的催化相、活性界面能够促进H 的分解/解离及H 的扩散,从而显著提升镁基材料的动力学性能。另外,金属与碳基复合催化剂对镁基材料的循环稳定性也有显著改善作用,可以作为一类理想的催化添加剂在未来进行更为深入的研究。
猜你喜欢
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
粉末冶金技术(2021年3期)2021-07-28
纤维复合材料(2018年3期)2018-04-25
中国有色金属学报(2018年2期)2018-03-26
电子测试(2017年11期)2017-12-15
信息记录材料(2016年4期)2016-03-11
焊接(2016年8期)2016-02-27
材料科学与工程学报(2016年5期)2016-02-27
应用化工(2014年10期)2014-08-16
