
顶空固相微萃取-气质联用法测定水中4种痕量环状缩醛类异嗅物质
2024-03-22 10:16:48狄欣宜
净水技术 2024年3期
狄欣宜
(上海城投原水有限公司,上海 200125)
饮用水嗅味是评价水质的重要感官指标。化工厂化学品泄漏及排放的工业废水是饮用水水源中异嗅味物质的常见来源。根据国外已有报道,异嗅物质2-乙基-4-甲基-1,3-二氧戊环(2-ethyl-4-methyl-1,3-dioxolane,2-EMD)和2-乙基-5,5-二甲基-1,3-二氧杂环乙烷(2-ethyl-5,5-dimethyl-1,3-dioxane,2-EDD)引发的污染事件在美国宾夕法尼亚州俄亥俄河、西班牙巴塞罗那、南美洲等地区皆有发生。这两种化合物大环中含有-CH2O-基团,为环状缩醛类化合物,两者被认为是树脂在酸催化聚合过程中二元醇所形成的副产物,经工业排放进入水体[1-3]。通过气味描述分析(FPA)发现,两种化合物的嗅阈值都在5~10 ng/L水平[4-5]。我国中科院生态环境研究中心在对全国98个水厂的调查中发现,2-EMD在水源水中的检出率为12.2%[6]。
目前,上海全市自来水原水已全部实现由水库集中取水,但现有四大饮用水水源地取水口位于长江和太湖流域下游,上游来水水质尚不稳定,突发性水污染事件时有发生,水源地取水口存在遭遇上游工业污水污染的可能。为了对上述环状缩醛类污染物构建有效的预警监测体系,需要对其建立高灵敏度的分析方法以适应低嗅阈值污染物的检测需要。
现有对2-EMD和2-EDD等环状缩醛类物质的检测主要通过闭环捕集法(CLSA)、液液萃取法(LLE)、顶空进样法(HS)、吹扫捕集法(P &T)、固相萃取法(SPE)对水样中的嗅味物质浓缩富集后,再由气相色谱法(GC-FID)、气相色谱质谱联用法(GC-MS)、气相色谱串联三重四级杆质谱法(GC-MS/MS)对待测物质进行分离与定性定量检测[1-3,7]。近年来,一种新型的顶空固相微萃取(HS-SPME)前处理技术逐渐为人所熟知,相较传统方法,顶空固相微萃取技术拥有萃取效率高、操作简单、无需使用溶剂萃取等诸多优势,已在水中2-甲基异莰醇、土臭素等藻源性嗅味物质的分析中拥有广泛应用。GC-MS/MS相较GC-FID和GC-MS拥有更强的抗背景干扰能力与更高的检测信噪比,但目前在实验室的普及率尚有限。本文研究了使用全自动顶空固相微萃取-气相色谱质谱联用法对2,2-二甲基-1,3-二氧环戊烷(22-DMD)、2-EMD、2-乙基-2-甲基-1,3-二氧戊环(2E2MD)、2-EDD 4种环状缩醛类物质进行痕量分析的可行性,同时对前处理条件、色谱与质谱的分析条件也进行了讨论和优化。
1 试验仪器、试剂与方法
1.1 仪器
仪器:Agilent 7890B/5977B气相色谱质谱联用仪,配备分流/不分流进样口,Extractor EI离子源(美国Agilent公司);Agilent PAL RSI自动样品前处理平台,配备孵化炉(Agitator)、SPME老化模块(美国Agilent公司)。
1.2 试剂和耗材
2-EMD、2-EDD混合标准品(2-EMD为顺反异构体混合物,顺反异构体浓度比例为58∶42,1 000 mg/L,1.2 mL,上海安普璀世标准技术服务有限公司);22-DMD标准品(≥98%,5 mL,上海安普试验科技股份有限公司);2E2MD标准品(≥98%,5 mL,上海安普试验科技股份有限公司);2-异丁基-3-甲氧基吡嗪(IBMP)标准溶液(100 mg/L,1 mL,美国o2si公司);氯化钠(优级纯,经450 ℃烘烤2 h后置于干燥器内备用);乙腈(色谱纯);超纯水(使用时现取);螺口顶空瓶(20 mL,配18 mm带隔垫螺口瓶盖);氦气(纯度≥99.999%)。
1.3 SPME参数设置
量取10.0 mL水样加入已称取2.0 g氯化钠的20 mL螺口顶空瓶中,在瓶中加入内标物IBMP后旋紧瓶盖并摇匀,将氯化钠充分溶解,放置于样品盘上等待测试。设置SPME孵化炉温度为60 ℃,转速为450 r/min,样品孵化5 min后萃取纤维在顶空瓶上方气相中顶空萃取25 min,萃取深度为22 mm,萃取完成后萃取纤维在气质联用仪进样口解吸5 min,进样深度为55 mm。萃取使用85 μm Carboxen/PDMS萃取纤维,新萃取纤维在进行分析前需在300 ℃下老化30 min,之后每次分析前在300 ℃下老化1 min。
1.4 GC-MS分析条件
色谱条件:色谱柱使用Agilent DB-5MS(60 m×0.25 mm×1.00 μm)石英毛细管柱,载气为氦气,恒流模式,柱流速为1.0 mL/min,不分流模式进样,进样口温度为250 ℃,程序升温设置为在35 ℃下保持2 min,然后以8 ℃/min的速率升至120 ℃保持2 min,最后以25 ℃/min的速率升至250 ℃保持9 min,传输线温度为280 ℃。
质谱条件:离子源EI源,离子源温度为230 ℃,离子化能量为70 eV,四级杆温度为150 ℃,扫描方式选择离子扫描(SIM),溶剂延迟9.0 min,选择离子参数如表1所示。

表1 化合物名称、保留时间、定量离子、定性离子
2 结果与讨论
2.1 色谱柱选择
在本文研究的4种环状缩醛类化合物中,2-EMD存在顺反异构体,2E2MD为2-EMD的同分异构体,3种化合物性质相似、色谱峰的保留时间相近。在气质联用分析中,对于保留时间相近、色谱峰存在重叠的不同化合物可以通过提取质谱离子化过程中形成的化合物的特征碎片离子并将其设置为化合物定性定量离子的方式使各化合物得以区分并准确定量。
经过对化合物NIST谱库质谱图的分析,适宜选作2-EMD定性定量离子的离子质荷比(m/z)为87、59、72,2E2MD为87、101、57。对于2-EMD,87离子峰为基峰,同时87也是2-EMD适宜选用的定性定量离子87、59、72中的最大值,受杂质干扰的可能性最小,因此87是2-EMD最适合的定量离子,但由于87离子峰同时也是2E2MD的基峰,如果使用87作为2-EMD的定量离子,2-EMD和2E2MD的色谱峰需要达到一定的分离度,否则2-EMD的定量将会受到2E2MD的干扰;对于2E2MD,87和101都是适合的定量离子,101离子峰的丰度小于87离子峰,101作为2E2MD定量离子时的检测灵敏度相对87为定量离子时的更小。
本试验测试了2-EMD和2E2MD的87选择离子色谱峰在部分水质分析中常用的色谱柱上的分离情况,测试结果表明,DB-WAX(30 m×0.25 mm×0.25 μm)能将难以分离的反式2-EMD和2E2MD完全分离,但顺式2-EMD另外会受到三氯甲烷的干扰,当使用DB-WAX(60 m×0.25 mm×0.25 μm)时,三氯甲烷与顺式2-EMD的87选择离子色谱峰依然高度重叠。三氯甲烷作为含氯消毒剂产生的主要消毒副产物在水厂出厂水及管网水中普遍存在,在水源水中也有部分检出,三氯甲烷对2-EMD的检测影响无法忽略,试验使用的DB-WAX固定相的色谱柱无法作为本试验的分析柱。试验另外测试了DB-624(60 m×0.25 mm×1.4 μm)和DB-5MS(60 m×0.25 mm×1.0 μm)两色谱柱对2-EMD和2E2MD的87选择离子色谱峰的分离效果,结果表明:若以分离度R≥1.5作为相邻两峰完全分离的评价标准,反式2-EMD与2E2MD在两种色谱柱上均无法得到完全的分离。根据美国EPA 524.2方法,在使用气质联用仪对水中挥发性有机物进行检测时,当拥有相似质谱图的同分异构体在色谱柱上无法完全分离,如果两异构体色谱峰重叠部分的峰谷高度在两异构体平均峰高的25%以下,则两峰的分离度可以接受,两种同分异构体可以分别定量,否则需将两异构体视为一组报告两者总量[8]。试验在上述两色谱柱使用优化后的升温程序对质量浓度为100 ng/L的2-EMD和2E2MD进行分离,结果显示反式2-EMD和2E2MD在两色谱柱上的分离情况满足EPA 524.2方法中的分离度要求,反式2-EMD和2E2MD可以分别报告定量结果。鉴于反式2-EMD和2E2MD未完全分离以及2-EMD除87外没有更适合的定量离子,本试验使用87作为2-EMD的定量离子,101作为2E2MD的定量离子。在试验过程中还发现,当使用DB-624固定相的色谱柱时,顺式2-EMD和反式2-EMD的87选择离子色谱峰另外会受到来自固相微萃取纤维本身的硅氧烷类物质六甲基环三硅氧烷的干扰。该干扰在样品顶空萃取完成后,萃取纤维进入气质联用仪进样口的解吸过程中随着纤维上被吸附的挥发性组分一同进入色谱柱,在每个样品中均稳定存在。虽然在DB-624(60 m×0.25 mm×1.4 μm)上该干扰可以通过程序升温设置将其出峰时间限制在两个2-EMD异构体的色谱峰之间,但其造成的基线抬升依然会对低浓度2-EMD样品的检测带来负面影响。考虑到两色谱柱对反式2-EMD与2E2MD的分离效果相近,本试验选择DB-5MS(60 m×0.25 mm×1.0 μm)为本文研究的4种环状缩醛类化合物的分析柱。
2.2 萃取条件的优化
2.2.1 萃取纤维及萃取温度的选择
固相微萃取技术中不同涂层材质的萃取纤维对不同化合物的吸附能力不同,萃取纤维的选择应从被分析物的分子质量、挥发性、极性、浓度级别等方面进行考量。本文分析的环状缩醛类化合物为痕量小分子的极性挥发性异味物质,试验比较了85 μm Carboxen/PDMS、50/30 μm DVB/CAR/PDMS、65 μm PDMS/DVB、85 μm Polyacrylate、100 μm PDMS 5种萃取纤维对4种环状缩醛类化合物的吸附效果,结果表明85 μm Carboxen/PDMS和50/30 μm DVB/CAR/PDMS这两种萃取纤维对目标环状缩醛类化合物的吸附效果较好。
在顶空分析中,平衡温度越高,被分析物在气液两相中的分配系数K越小,顶空中被分析物的含量越大,检测灵敏度越高。但对于顶空SPME分析,被分析物在顶空与萃取纤维的涂层中存在新的分配。同时,对水分析而言,平衡温度越高,萃取纤维涂层吸收的水分越多,对后续的分析仪器与检测结果也会产生一定影响。为确认不同萃取纤维对环状缩醛类化合物的最佳平衡温度,本试验对85 μm Carboxen/PDMS和50/30 μm DVB/CAR/PDMS两种萃取纤维在40~70 ℃平衡温度下4种环状缩醛类化合物的峰面积响应进行了比较,结果如图1~图2所示。85 μm Carboxen/PDMS萃取纤维相对50/30 μm DVB/CAR/PDMS萃取纤维获得的峰面积响应更高,除2-EDD的响应随温度的升高而升高外,其余3种化合物都在60 ℃有最高响应,考虑到平衡温度高时萃取纤维吸附的水分的影响,本试验选择使用85 μm Carboxen/PDMS萃取纤维,以60 ℃作为4种环状缩醛类化合物的平衡温度。
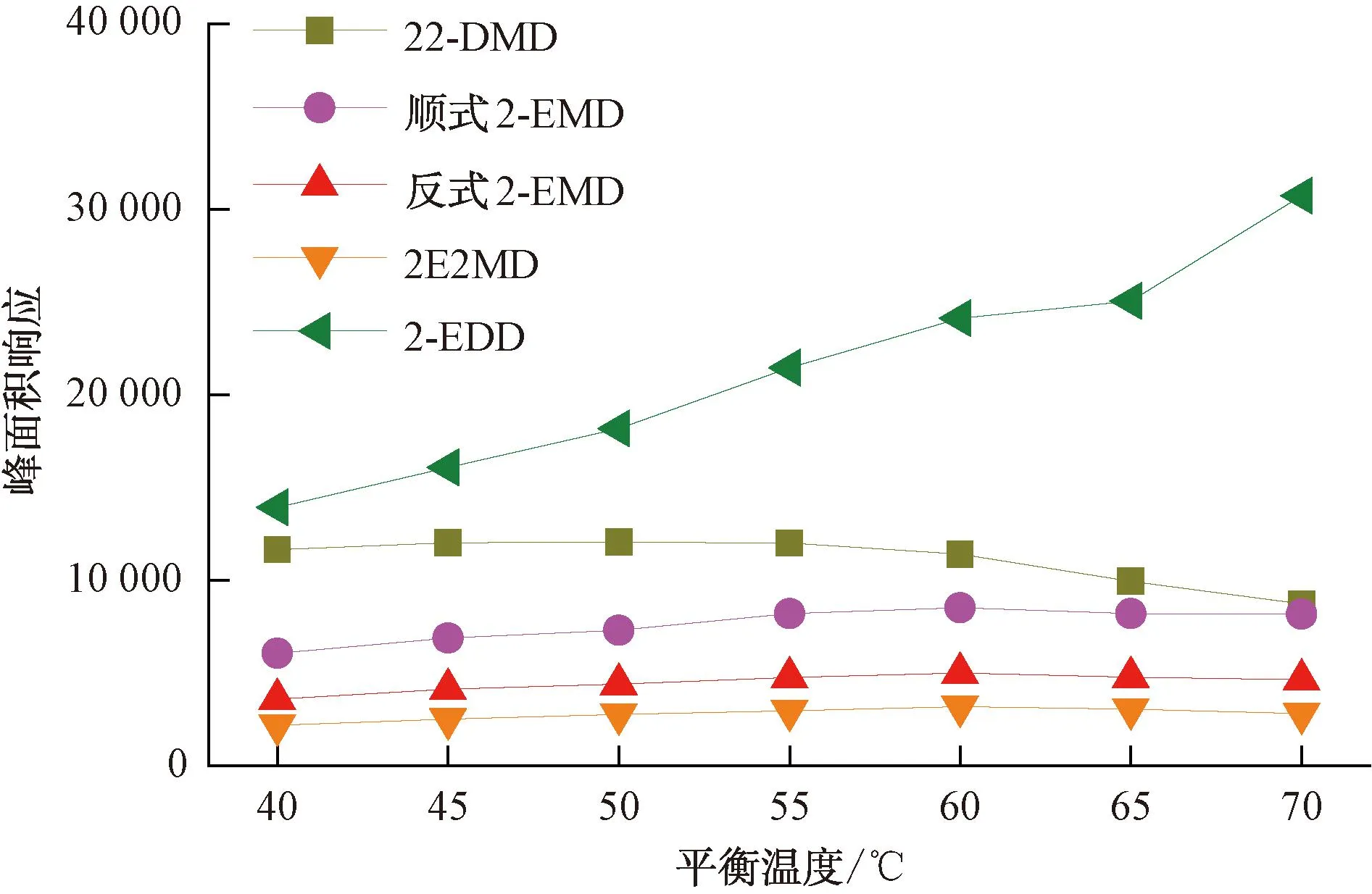
图1 85 μm Carboxen/PDMS萃取纤维在不同平衡温度下目标化合物的峰面积响应变化

图2 50/30 μm DVB/CAR/PDMS萃取纤维在不同平衡温度下目标化合物的峰面积响应变化
2.2.2 样品体积、萃取深度、孵化时间及震摇速度的选择
顶空分析中,对于固定容积的顶空瓶,更大的取样量对应更低的相比率β,更低的相比率通常会使检测的灵敏度增加。本试验使用20 mL容积顶空瓶,取样量为10 mL,萃取纤维为1 cm长度的85 μm Carboxen/PDMS,对应的萃取深度为22 mm,孵化时间设置为5 min。
本试验测试了孵化器不同振摇速度对目标化合物的峰面积响应的影响,结果如图3所示。振摇速度达到450 r/min后,4种环状缩醛类化合物的峰面积响应不再有显著变化,本试验选择450 r/min作为孵化器的振摇速度。

图3 不同孵化器振摇速度对目标化合物的峰面积响应影响
2.2.3 盐析作用的影响
顶空分析中,利用盐析作用即在水溶液中加入无机盐以降低被分析物在气液两相中的分配系数K也是提高顶空气相中被分析物含量的有效手段。本试验使用氯化钠为盐析剂,测试了不同添加量的氯化钠对目标化合物峰面积响应的影响,结果如图4所示。4种环状缩醛类化合物的峰面积响应均随着氯化钠添加量的增加而增加。当取样量为10 mL时,考虑到氯化钠添加量对溶液体积变化的影响及日常分析中氯化钠在水中溶解的难易程度,本试验在10 mL水样中添加2.0 g氯化钠,即水样中添加的氯化钠的质量分数为20%。

图4 不同氯化钠添加量对目标化合物的峰面积响应影响
2.2.4 进样深度的选择
试验发现,萃取纤维在进样口解吸时的进样深度对目标化合物的峰型及峰面积响应也会产生影响。当进样深度较浅,不分流进样的低载气流量造成了待测组分在进样口的扩散,少量组分随后从分流出口流出造成损失,峰面积响应也随之降低。本试验比较了4种目标化合物在不同进样深度下的峰面积响应,结果如图5所示。当进样深度达到55 mm时,目标化合物的峰面积响应不再随进样深度的增加而发生显著变化。本试验选择55 mm为解吸时的进样深度。

图5 不同进样深度对目标化合物的峰面积响应影响
2.2.5 萃取时间的选择
在顶空SPME分析中,被分析物存在气液两相平衡及顶空气相与萃取纤维涂层之间的平衡两组平衡分配,当萃取纤维对顶空气相中的挥发性组分萃取一定时间后,吸附在萃取纤维上的被分析物含量将达到相对恒定值。本试验研究了60 ℃的平衡温度下,不同的萃取时间对4种环状缩醛类化合物的峰面积响应影响,结果如图6所示。对于22-DMD、2-EMD、2E2MD 3种化合物,当萃取时间超过25 min时,峰面积响应不再显著增加,萃取在25 min时基本达到平衡;对于2-EDD,萃取在35 min时基本达到平衡。综合考虑试验效率及长时间萃取造成的萃取纤维中水分增多的影响,本试验选择的萃取时间为25 min。

图6 不同萃取时间对目标化合物的峰面积响应影响
2.3 内标物选择
试验比较了4种目标化合物与氟苯、氯苯-d5、1,2-二氯苯-d4、IBMP 4种内标物在前述2.2.3小节和2.2.5小节中的峰面积响应变化情况,结果如图7、图8所示。相对于氟苯、氯苯-d5及1,2-二氯苯-d4,IBMP与4种环状缩醛类化合物的峰面积响应变化趋势更趋于一致,在试验条件发生变化时目标化合物与IBMP的峰面积响应比值能相对保持恒定,从而将试验条件的变化对检测带来的影响降至最低,故本试验选择使用IBMP为内标物。

图7 目标化合物和内标物的峰面积响应在盐析作用影响下的变化

图8 目标化合物和内标物的峰面积响应在不同萃取时间下的变化
2.4 方法评价
2.4.1 标准曲线
配制22-DMD质量浓度为25、50、125、250、375、500 ng/L,2-EMD、2E2MD、2-EDD质量浓度为5、10、25、50、75、100 ng/L,内标物IBMP质量浓度为20 ng/L的标准溶液系列,按照优化后的条件将上述标准溶液按浓度由低至高的顺序依次进样分析。目标化合物与内标物的色谱图及各化合物定量离子色谱图如图9~图14所示。

图9 TIC图(SIM方式)(22-DMD,2-EMD,2E2MD,2-EDD,2BMP分别为500,100,100,100,20 ng/L)

图10 500 ng/L 22-DMD定量离子色谱图

图11 100 ng/L 2-EMD定量离子色谱图

图12 100 ng/L 2E2MD定量离子色谱图

图13 100 ng/L 2-EDD定量离子色谱图

图14 20 ng/L IBMP定量离子色谱图
以标准溶液系列中目标化合物与内标物的浓度比值为横坐标,峰面积响应比值为纵坐标,用最小二乘法建立校准曲线,曲线的线性相关系数r均大于0.995,曲线在对应的浓度范围内线性良好。各目标化合物的校准曲线方程、线性相关系数、线性范围如表2所示。

表2 4种环状缩醛类化合物校准曲线方程及相关参数
2.4.2 方法检出限与测定下限
根据《环境监测分析方法标准制订技术导则》(HJ 168—2020)附录A中对方法检出限的确定方法[9],按优化后的试验条件平行测定7次加标浓度为估计方法检出限值3~5倍的纯水加标样品,实际配制的加标溶液质量浓度为22-DMD 5 ng/L、2-EMD 5 ng/L、2E2MD 5 ng/L、2-EDD 1 ng/L、内标物IBMP 20 ng/L,检测后计算得到的各目标化合物的方法检出限与测定下限如表2所示,4种环状缩醛类化合物的检出限为0.3~1.6 ng/L,能够满足痕量级浓度水平样品的检测需要。
2.4.3 方法精密度与准确度
为评价方法的精密度与准确度,按优化后的方法对纯水、水源水、水厂出厂水和管网水水样分别进行低、中、高3种不同浓度的加标试验,不同本底和加标浓度各平行测定6次。加标质量浓度除22-DMD为50、250、500 ng/L外,2-EMD、2E2MD、2-EDD均为10、50、100 ng/L,内标物IBMP质量浓度均为20 ng/L。加标试验中使用的水源水、水厂出厂水和管网水水样中4种环状缩醛类化合物的浓度均小于方法检出限。各目标化合物的相对标准偏差和回收率结果如表3~表6所示。纯水、水源水、水厂出厂水和管网水加标后测得的目标化合物的相对标准偏差分别为1.8%~8.2%、2.0%~8.5%、2.1%~7.5%和1.6%~8.6%,回收率分别为94%~115%、101%~110%、98%~117%和99%~114%,方法精密度和准确度能够满足质量控制要求。

表3 纯水中4种环状缩醛类化合物加标试验结果

表4 原水中4种环状缩醛类化合物加标试验结果

表5 水厂出厂水中4种环状缩醛类化合物加标试验结果

表6 管网水中4种环状缩醛类化合物加标试验结果
2.5 方法应用
按优化后的方法对长江和太湖流域下游水源中的4种环状缩醛类化合物浓度进行测定,测定结果如表7所示。

表7 原水中4种环状缩醛类化合物浓度测定结果
3 结论
本文采用顶空固相微萃取-气质联用技术,建立了水中4种痕量环状缩醛类异嗅物质的检测方法。固相微萃取前处理过程使用85 μm Carboxen/PDMS萃取纤维,水样在60 ℃下以450 r/min的速度孵化5 min后,顶空萃取25 min,盐析剂氯化钠添加的质量分数为20%,色谱柱使用Agilent DB-5MS石英毛细管柱(60 m×0.25 mm×1.00 μm),内标物使用IBMP。
22-DMD在25~500 ng/L,2-EMD、2E2MD、2-EDD在5~100 ng/L的质量浓度内线性相关系数r均大于0.995,目标化合物的方法检出限在0.3~1.6 ng/L。纯水、水源水、水厂出厂水和管网水平行加标样品中目标化合物的相对标准偏差为1.6%~8.6%,回收率为94%~117%。本方法能够满足水中4种痕量环状缩醛类异嗅物质的测定需要。
猜你喜欢
现代实用医学(2022年10期)2022-12-08 05:50:08
大学化学(2022年1期)2022-02-28 12:16:50
中国民间疗法(2021年9期)2021-07-22 08:05:52
合成树脂及塑料(2020年1期)2020-03-27 14:14:02
中成药(2018年12期)2018-12-29 12:26:08
化工管理(2017年12期)2017-03-04 00:07:45
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:36:55
合成化学(2015年1期)2016-01-17 08:55:47
天然产物研究与开发(2014年7期)2014-04-27 14:16:08
组合机床与自动化加工技术(2014年12期)2014-03-01 02:22:47
