
活性炭强化泡沫钛基空气扩散电极电过臭氧氧化及降解布洛芬效能
2024-03-18 05:55:00刘思蓓梅竣乔谢谨裕刘轶君邓凤霞
应用化学 2024年2期
刘思蓓 梅竣乔 谢谨裕 刘轶君 邓凤霞* 邱 珊*
1(哈尔滨工业大学环境学院,哈尔滨 150000)
2(成都市排水有限责任公司,成都 610023)
新污染物是指对生态环境或人体健康存在风险,尚未纳入管理或者现有管理措施不足以有效防控其风险的污染物,由于其隐蔽性、持久性、危害程度大以及治理困难等特点,已成为生态环境保护工作的重点防控对象[1]。布洛芬作为一种广泛检测到的新污染物,通过传统污水处理技术也难以有效去除,且无法在人体内完全代谢,在环境中存在潜在风险[2-4]。因此,需要开发出高效去除布洛芬的工艺。
电过臭氧(Electro-peroxone)技术作为一种基于臭氧的新型高级氧化废水处理技术,在pH 值为3.00~10.00的范围内具有良好的处理效果。该技术将臭氧氧化与电化学技术结合,阴极经由二电子路径将氧气(O2)还原为过氧化氢(H2O2),生成的H2O2进一步与溶液中的臭氧(O3)发生反应产生氧化性强的羟基自由基(HO•)降解污染物,具体反应过程可由式(1)—(9)描述。该技术具有消毒副产物较少、无需存储和运输H2O2等优点,发展潜力巨大[5-7]。与传统废水处理工艺相比,电过臭氧技术具有更为便捷、安全、经济和节能等优势,已用于新污染物的处理,并取得了良好的效果[6,8]。例如,Wa等[9]对比常规臭氧技术、过臭技术和电过臭氧技术在不同水体中降解布洛芬等耐臭氧药物的效能,表明E-peroxone工艺能更明显地促进O3向HO•的转化,提高去除效率。Li等[10]对常规臭氧氧化和电过臭氧工艺降解布洛芬情况进行了比较,结果表明,电过臭氧技术在5~15 min就可以使布洛芬完全降解,其速度远远快于臭氧氧化(≥30 min)过程。此外,Huber 等[11]研究提到在4 种自然水体中,布洛芬的降解率可以从臭氧氧化的24%~68%提高到电过臭氧处理的78%~90%。与常规的臭氧氧化相比,电过臭氧主要通过形成的HO•来氧化有机物,布洛芬与臭氧直接氧化的二级速率常数仅为9.6 L/(mol·s),矿化效果较差,但布洛芬与HO•反应的二级速率常数高达7.4×109L/(mol·s)[10],因此,可以借助电过臭氧技术对布洛芬进行降解处理。
电过臭氧技术在降解布洛芬方面展现出独特的优势,但布洛芬降解效率的提高依旧是一大问题,主要归因于以下2个方面:首先,由于电极尺寸和曝气装置的影响,反应器中存在氧气传质困难、臭氧利用率低以及电流密度较小等问题[7];其次,由于氧气、极板和电解质等非均相物质之间的气态反应物传质受限,导致原位H2O2产生速率受到限制,进而阻碍了HO•的产生速度,而HO•作为布洛芬降解的主要活性物质,会导致其降解效率降低[6]。
本研究采用微孔泡沫钛空气扩散电极填充活性炭构建(AC@Ti-F GDE)电过臭氧体系,通过与3 种不同体系对比,探究传质方式和活性炭投加方式对HO•、液相臭氧浓度(O3,liquid)和H2O2等活性物质含量影响机制,并进一步通过测定HO•相对含量、H2O2浓度和物化表征探究活性炭投加量和颗粒大小等活性炭参数对强化O3催化和H2O2的生成作用和分解作用进行机理剖析。然后,将该体系应用于布洛芬降解,探究pH 值、电流密度和臭氧浓度不同因素对布洛芬降解效果的影响,得到最佳反应参数。最后,通过测定活性物质及其贡献程度,降解过程中的总有机碳(TOC)以及利用液相色谱质谱等技术,推导其降解矿化机理和可能存在的降解路径。
1 实验部分
1.1 仪器和试剂
T10-3S 型臭氧发生器(北京同林臭氧公司);GM-6000-OEM 型臭氧检测器(德国Ansero 公司);Smart2Pure3 型超纯水仪(美国Thermo Fisher Scientific 公司);ACQUIT UPLC 型超高效液相色谱(HPLC,美国Waters公司);UPLC-Q-TOF型液相色谱-质谱联用仪(LC-MS,美国Agilent公司);IRAffinity-1型傅里叶红外光谱仪(FT-IR,日本Shimadzu 公司);CHI660E 型电化学工作站(上海辰华公司);RRDE-3A 型旋转环盘电极(RRDE,武汉高仕睿联科技有限公司);TOC-VCPN 型TOC 测定仪(日本Shimadzu公司);D8 ADVNCE 型X 射线衍射仪(XRD,德国Bruker 公司);Kalpha 型X 射线光电子能谱仪(XPS,美国Thermo Fisher Scientific 公司);EMXplus 型电子自旋共振光谱仪(ESR,德国Bruker 公司);ZEISS SIGMA 500 型扫描电子显微镜(SEM,美国Ted Pella 公司);2 cm×3 cm 硼掺杂金刚石电极(BDD 电极,上海越磁电子科技有限公司);2 cm×3 cm铂片电极(上海越磁电子科技有限公司);C18液相色谱柱(美国Agilent公司)。
无水硫酸钠(分析纯)、氢氧化钠(分析纯)、浓硫酸(分析纯)、无水乙醇(分析纯)购自国药集团化学试剂公司;活性炭、对苯二甲酸(分析纯)、5,5-二甲基-1-吡咯啉-N-氧化物(DMPO,分析纯)、2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO,分析纯)购自上海阿拉丁试剂有限公司;无水硫酸钠(分析纯)、浓盐酸(分析纯)、碘化钾(分析纯)、邻苯二甲酸氢钾(分析纯)、硫代硫酸钠(分析纯)、磷酸二氢钠(分析纯)、磷酸氢二钠(分析纯)、异丙醇(分析纯)购自西陇科学股份有限公司;布洛芬(分析纯)、靛红钾(分析纯)购自上海麦克林生化科技有限公司;Nafoin(质量分数5%)购自Sigma-Aldrich 公司;甲醇(色谱纯)、甲酸(色谱纯)、乙腈(色谱纯)购自北京DIKMA公司。
1.2 实验装置
反应装置示意图如图1所示,主要组成为:内径为70 mm,高度为80 mm的圆柱形玻璃反应器,配备密封式聚四氟乙烯盖(盖上有4个开孔M12-6.7、1个开孔为M12-8.5和1个M6-3.2的取样口)。在测定H2O2时,阳极采用硼掺杂金刚石(BDD)电极,阴极采用AC@Ti-F GDE;而在测定污染物降解时,阳极采用铂片电极,阴极采用AC@Ti-F GDE。所有实验均使用200 mL 50 mmol/L 的Na2SO4作为电解质,实验过程均采用恒电流模式,通过控制电源的输出来进行实验。目前,在国内外污水处理厂进水中布洛芬最高检出质量浓度(ρ)如表1所示[9-13],故设置目标污染物布洛芬初始质量浓度为10 mg/L。

表1 不同污水处理厂进水中布洛芬检出最高质量浓度Table 1 The highest mass concentration of ibuprofen detected in influent of different sewage treatment plants

图1 电过臭氧反应装置图(A)及设计的电极结构(B)Fig.1 Drawing (A) and design of electrode structure (B) of electro-peroxone reaction equipment
泡沫钛空气扩散电极Ti-F GDE 的结构如图1B 所示,电极由长为103.6 mm,直径为6 mm 的曝气管和长为24.1 mm,直径为31 mm,体积为12.1 cm3的半椭球状的曝气头组成,曝气头上曝气孔径为10 μm。而AC@Ti-F GDE的制备是在Ti-F GDE的基础上添加一定量活性炭颗粒得到的,当活性炭投加的质量分别为10、100和150 mg时,得到活性炭投加密度为0.8、8.3和12.4 mg/cm3的体系进行研究。
1.3 分析测试方法
1.3.1 HO•的测定
采用对苯二甲酸(TA)作为对HO•的捕获剂,并通过荧光分光光度法进行测定[17-18]。在实验过程中,每隔3 min 取1.00 mL 电解质,总共测定15 min。TA-OH 的激发波长设置为315 nm,发射波长设置为429 nm,扫描范围限定在350~480 nm之间。扫描速度设定为1500 nm/min。通过监测在429 nm处的荧光强度变化来评估体系中HO•的相对含量变化[19]。
1.3.2 H2O2的测定
使用碘化钾分光光度法进行测定[17],在15 min 内,每隔3 min,取出1.00 mL 电解质,将其分别转移到10.00 mL比色管中。随后,依次加入1.00 mL的邻苯二甲酸氢钾溶液(浓度为0.1000 mol/L)和1.00 mL碘化物溶液。样品经过超纯水稀释定容后,进行充分摇匀,并静置10 min。之后,在波长为351 nm 的条件下,利用1 cm 比色皿测定其吸光度。通过H2O2浓度和吸光度之间的曲线方程:y=0.1034x+0.0019(其中,x为吸光度,y为H2O2浓度(mmol/L)),计算H2O2的浓度。
1.3.3 布洛芬的测定
15 min 内,每隔3 min,取1.00 mL 电解质。随后,向每个样品中加入50 μL 的0.1000 mol/L 硫代硫酸钠来终止反应。过滤之后,将反应混合液转移到色谱小瓶中。采用HPLC 测定反应过程中布洛芬的浓度。使用C18液相色谱柱,柱温设置为30 ℃。在UV检测器中,设置特征波长λ为220 nm。流动相的组成为60%乙腈和40%超纯水(含有体积分数0.01%甲酸)。进样的体积为5 μL。设定流速为0.1 mL/min,测试时间为5 min。
1.3.4 臭氧的测定
采用靛红钾法对水中的臭氧进行吸光度测定。臭氧与靛红钾反应可以使靛红钾脱色,且反应速度极快,产物稳定,因此可以通过分光光度法对臭氧进行测定[20]。首先,取6 个10.00 mL 的容量瓶,每个容量瓶加入1 mL 磷酸缓冲溶液(将28 g NaH2PO4和35 g H3PO4溶于蒸馏水中并稀释至1 L来配置缓冲溶液)。随后,向每个容量瓶中加入0.50 mL 靛红钾溶液。其中,一个容量瓶被选作空白样品,直接用蒸馏水稀释至刻度。其它容量瓶在15 min 内,每隔3 min 取出0.50 mL 含有臭氧的水样,加入适量的蒸馏水定容。经充分摇匀后,使用分光光度计在波长λ=610 nm 进行比色分析。按照下述公式(10)计算臭氧的液相质量浓度。
式中,ρ为液相臭氧质量浓度(mg/L),A0和A1分别为通入臭氧前后的溶液吸光值,48×103为臭氧摩尔质量(mg/mol),ε为靛红钾摩尔吸光值(20400 L/(mol·cm)),L为比色皿厚度(cm),VTotal和VO3分别为含臭氧前后的溶液的总体积与体积(mL)。
1.3.5 布洛芬降解产物测定
采用LC-MS 测定布洛芬降解过程中产生的中间产物。流动相的流速为0.2 mL/min。采用梯度洗脱法,在15 min 内,甲醇和体积分数0.1%甲酸的体积比从35∶65 逐渐增加到80∶20,并保持此比例运行5 min。质谱部分采用负离子模式,并扫描范围设定为50~750m/z[21]。
1.4 旋转环盘电极(RRDE)分析
采用RRDE来测量氧饱和电解质中填充了不同颗粒活性炭后的二电子ORR选择性的电子转移数(n)。RRDE测试中,使用AC@Ti-F GDE 作为工作电极,铂片作为阳极,饱和甘汞电极(0.1 mol/L KOH)作为参比电极。在进行测量之前,通过在1.3~0 V 范围内以10 mV/s 的扫描速率对工作电极进行循环伏安洗涤,直到在N2饱和电解质条件下获得稳定的循环伏安曲线。随后,以1600 r/min的旋转速度进行氧还原反应的线性扫描伏安法,通过在1.2 V 处测量分解环电流来确定H2O2的生成量。将催化剂(5 mg)混合到异丙醇(50 μL)、水(450 μL)和Nafion 溶液(50 μL)中制备电催化剂墨水。溶液超声处理以获得均匀分散的溶液,然后将10 μL 墨水滴在抛光的洁净玻璃碳工作电极上,红外干燥。使用[Fe(CN)6]4-/[Fe(CN)6]3-氧化还原体系在N2饱和电解质溶液中测定环盘工作电极的收集效率(N)。根据公式(11)计算ORR反应的n。
式中,n为电子转移数,Idisk、Iring分别为圆盘电流和环形电流(mA),N为环盘电极的收集效率,0.32。
2 结果与讨论
2.1 不同电过臭氧体系效能
对4种电过臭氧体系效能进行研究,分别为传统的阴极曝气的体系(Ti-F体系)、阴极曝气但未填充活性炭的气体扩散电极体系(Ti-F GDE 体系)、活性炭填充于阴极内(AC@Ti-F GDE 体系)和活性炭直接投加至电解质溶液(AC+Ti-F GDE 体系)的体系。通过对比Ti-F 和Ti-F GDE 体系来研究阴极微气泡曝气和三相界面构建对活性物质生成的影响,对比AC@Ti-F GDE 和AC+Ti-F GDE 体系来研究活性炭投加方式对活性物质生成的影响。
探究了4种电过臭氧体系羟基自由基产量、液相臭氧浓度和H2O2积累量的不同,结果如图2所示。在反应体系中,HO•的相对含量从高到低排序为:AC@Ti-F GDE>AC+Ti-F GDE>Ti-F GDE>Ti-F。Ti-F体系的HO•含量远低于其它体系,这是因为其它体系采用微气泡方式曝气,而Ti-F体系采用大气泡方式曝气,影响了氧气的传质,从而限制了ORR 反应,减少了HO•的生成。AC+Ti-F GDE 体系的HO•含量仅略高于Ti-F GDE 体系,远低于AC@Ti-F GDE 体系,这是因为直接将活性炭投加到溶液中的方式降低了活性炭与阴极的碰撞几率,影响了H2O2的生成。因此,采用荧光光度法测定的微孔泡沫钛空气扩散电极(Ti-F GDE)和传统的电极(Ti-F)体系HO•的荧光强度由2517 a.u 增加到6436 a.u,其相对含量提升155.7%。当在Ti-F GDE 中填充粒径为850 μm、密度为0.8 mg/cm3的活性炭后,AC@Ti-F GDE 体系中测得的HO•的荧光强度为8716 a.u,HO•的相对含量相较于Ti-F GDE体系进一步提升了35.4%。

图2 不同体系HO•相对含量(A)、O3,liquid浓度(B) 和H2O2的积累量 (C)。反应在电流为50 mA,活性炭粒径为850 μm,投加密度为0.8 mg/cm3,Na2SO4溶液浓度为50 mmol/L,pH 值为7,O3,gas质量浓度为16.5 mg/L,曝气流量为50 mL/min,反应时间为15 min的条件下进行Fig. 2 Relative content of HO• (A),concentration of O3,liquid (B) and accumulation of H2O2 (C) in different systems.The reaction was carried out under the conditions of current of 50 mA,activated carbon particle size of 850 μm,dosage of 0.8 mg/cm3,Na2SO4 solution concentration of 50 mmol/L,pH of 7,O3,gas mass concentration of 16.5 mg/L,aeration flow rate of 50 mL/min and reaction time of 15 min
O3,liquid在3 min 内迅速达到相对稳定的状态。不同体系的O3浓度由于O3的分解而在一定范围内出现正常波动,从高到低排序为:Ti-F GDE>AC+Ti-F GDE>AC@Ti-F GDE>Ti-F。AC+Ti-F GDE 和AC@Ti-F GDE 体系由于投加活性炭,可以催化O3的分解生成HO•,所以尽管O3浓度值低于Ti-F GDE 体系,但它们的HO•含量仍然高于Ti-F GDE 体系。而在AC@Ti-F GDE 体系中,活性炭在电极内部的局部区域保持流化状态,导致部分曝气孔径堵塞,进一步微弱地影响了O3的传质和催化效果[22]。
气、固、液三相界面和微气泡曝气方式促进了反应物传质,活性炭填充阴极内部,提高了与阴极的碰撞几率,形成微电场,促进了ORR反应生成H2O2,而活性炭直接投加于电解质溶液中的方式,可能由于在磁力搅拌的作用下与阴极碰撞几率减小或者其可以促进生成的H2O2分解为HO•,导致了该体系H2O2积累量有所减少。因此,反应体系H2O2积累量从高到底排序为:AC@Ti-F GDE>Ti-F GDE>AC+Ti-F GDE>Ti-F。
综上,AC@Ti-F GDE 电过臭氧体系具有优异效能的原因主要有3 个方面:1)AC@Ti-F GDE 可通过建立气、固、液三相界面和微气泡曝气方式促进O2传质,所填充的活性炭可提高二电子ORR反应活性和选择性,强化了H2O2积累,促进H2O2与O3发生过臭反应生成大量的HO•;2)采用微气泡曝气方式和填充活性炭可催化H2O2和O3,促进了HO•等活性物质生成;3)微气泡曝气促进O3液相传质。
2.2 AC@Ti-F GDE体系强化臭氧催化参数
2.2.1 活性炭投加密度
图3 展示了在活性炭投加密度分别为0、0.8、8.3 和12.4 mg/cm3时,通过荧光分光光度法测得的体系在0、3、6、9、12 和15 min 时的HO•相对含量。结果表明,活性炭的投加可以提高HO•相对含量,进而证实了活性炭投加能够促进O3的催化反应,提高O3的利用效率。在反应前3 min 内,随着活性炭投加密度的增加,HO•相对含量逐渐增加。然而,在3 min 后,HO•相对含量的变化不大。经过反应15 min 后,体系中的HO•相对含量从高到低依次为0.8、8.3 和12.4 mg/cm3。这一现象可能是由于在前3 min内,臭氧催化反应受到活性炭位点数量的限制,因此随着投加密度的增加,HO•相对含量增加。而在3 min 后,随着活性炭投加密度的增加,部分曝气孔发生局部沉降堵塞,导致单个曝气孔的流速增大,微气泡从曝气口脱离并减少在电解液中停留的时间,从而阻碍了O3的传质,进而影响了HO•相对含量。因此,对于AC@Ti-F GDE 臭氧曝气体系,最优的活性炭投加密度为0.8 mg/cm3。

图3 活性炭投加密度为0、0.8、8.3和12.4 mg/cm3对HO•相对含量影响Fig.3 Effect of activated carbon dosage density of 0,0.8,8.3 and 12.4 mg/cm3 on the relative content of HO•.No current application; d(GAC)=850 μm; c(Na2SO4)=50 mmol/L; pH=7; Reration flow rate=50 mL/min;O3,gas=15 mg/L; reaction time=15 min
2.2.2 活性炭颗粒粒径
图4 展示了采用荧光分光光度法测得的活性炭颗粒粒径为75、150 和850 μm 的AC@Ti-F GDE臭氧曝气体系,在反应15 min 后的HO•相对含量。一些研究表明,较小的颗粒粒径对应着更大的比表面积,可以促进O3的催化氧化反应[23-24]。然而,随着颗粒粒径的增大,HO•相对含量逐渐增加。这可能是由于较大的活性炭颗粒粒径导致传质阻力下降,从而促进了O3的催化氧化反应[24-25]。因此,提出AC@Ti-F GDE 臭氧曝气体系中活性炭颗粒粒径主要通过影响O3的传质阻力来强化O3的催化氧化反应,对比表面积的影响要弱于对O3传质阻力的影响。因此,最佳的活性炭粒径为850 μm。

图4 不同活性炭颗粒粒径对于HO•相对含量影响Fig. 4 Effect of different activated carbon particle diameters on HO• relative content
2.3 AC@Ti-F GDE体系强化H2O2积累参数
2.3.1 活性炭投加密度
通过实验研究了不同活性炭投加密度(0、0.8、8.3和12.4 mg/cm³)对H2O2积累量的影响,结果显示,随着活性炭投加密度的增加,H2O2积累量逐渐减少(图5A)。这一结果可以解释为,活性炭与阴极之间的接触时间受到活性炭投加密度的影响[26]。在实验过程中,随着活性炭投加密度的增加,虽然活性炭与阴极之间碰撞频率增加,但减少了与阴极接触时间,降低了H2O2产量。此外,其它研究也表明活性炭投加密度的增加会增加H2O2分解的活性位点,进一步加剧H2O2的分解,从而减少其积累量[27-28]。

图5 不同活性炭投加密度(A)及电流(B)对H2O2的积累量影响Fig.5 Effect of different activated carbon dosage density (A) and current (B) on the accumulation of H2O2 Reaction current:50 mA;100 mA.Particlesize:850 μm; c(Na2SO4)=50 mmol/L; pH=7; v(oxygen aeration)=50 mL/min;Anode:BDD,reaction time≥15 min
为了进一步验证活性炭投加密度对H2O2积累的影响机制,将电流从50 mA 提高到100 mA,增加活性炭与阴极碰撞获得的电子数量,并研究H2O2积累量的变化趋势(图5B)。结果显示,随着电流的增加,不同活性炭投加密度的体系中H2O2积累量均有所增加。然而,在电流为50 mA 时H2O2积累量的从高到低顺序依次对应活性炭投加密度为0.8、8.3 和12.4 mg/cm³时,电流为100 mA 时其顺序变为8.3、0.8和12.4 mg/cm³。这说明即使活性炭投加密度增加导致接触时间变短,但电流的提高加快了单位时间内的电子速率。这一结果进一步验证了活性炭投加密度的变化会影响活性炭获得电子的能力,从而影响H2O2的积累。
2.3.2 活性炭颗粒粒径
活性炭粒径大小对其催化性能影响巨大。为此,探究了活性炭颗粒粒径分别为75、150 和850 μm时对AC@Ti-F GDE臭氧曝气体系H2O2积累量的影响,结果如图6A所示。随着活性炭颗粒粒径的增加,H2O2的积累量呈现先降低后增加的趋势,反应15 min 后,75、150 和850 μm 的体系H2O2的积累量分别为26.8、25.5和32.6 μmol/L。活性炭颗粒粒径对H2O2积累量的影响主要取决于:1)不同粒径的活性炭催化分解H2O2的能力不同;2)不同粒径活性炭生成H2O2能力不同,下面将进行进一步讨论。

图6 不同粒径活性炭对H2O2积累量(A)和H2O2分解(B)的影响及其吸附等温线(C)和孔径分布图(D)Fig. 6 Effects of different particle sizes activated carbon on the accumulation (A) and decomposition (B) of H2O2,as well as their adsorption isotherms (C) and pore size distribution (D)
首先,讨论不同粒径下H2O2的分解作用。对H2O2的自分解作用、Ti-F 电极的H2O2分解作用以及不同粒径活性炭对H2O2的催化分解作用进行了探究。结果如图6B 所示,Ti-F 电极的H2O2分解作用相对较弱,而随着活性炭颗粒粒径的增大,H2O2的分解作用增强。因此,与75 μm 的活性炭相比,150 μm 的活性炭具有较低的H2O2积累量。
催化剂的性能与其孔径分布息息相关。为此,对不同粒径的活性炭进行了BET 测试,得到了其吸附曲线(图6C),并利用密度泛函理论计算获得了孔径分布图(图6D)。结果表明,850 μm的活性炭呈现典型的Ⅰ型吸附等温线,表明存在微孔填充现象;而150 和75 μm 的活性炭则属于Ⅱ型吸附等温线,表明存在中孔和大孔结构。孔径分布图也显示,850 μm 的活性炭相比150 和75 μm 的活性炭具有更高的微孔占比。相关研究已经表明,微孔为主的活性炭导致了H2O2和O3的停留时间延长[29-30],从而加剧了H2O2的分解转化为H2O和O3分解为HO•等活性物质的过程。
其次,研究了不同粒径下H2O2的生成作用。H2O2通过二电子ORR 产生,为探究活性炭颗粒粒径对ORR选择性的影响,通过RRDE测试计算其电子转移数,结果如图7所示。具体而言,75、150和850 μm颗粒粒径的活性炭电子转移数分别为2.6、2.3和2.1,表明不同粒径活性炭的二电子选择性从优到差依次为850、150和75 μm。

图7 不同粒径活性炭的RRDE图(A)及电子转移数(B)Fig. 7 Rotating ring-disk electrode (RRDE) diagram (A) and electron transfer number (B) of activated carbon with different particle sizes
为了进一步研究颗粒粒径越大导致电催化活性增强的原因,进行了XRD 分析以获得不同粒径活性炭的原子排列情况,如图8A 所示。在25 和43(°)附近观察到衍射峰,为石墨中(002)和(100)平面对应的峰,表明活性炭具有石墨结构[31]。值得注意的是,850 μm 的活性炭相较于75 和150 μm 的活性炭,衍射峰更高,这意味着其石墨化程度更高,电阻率更低。接着,采用拉曼光谱进一步研究了活性炭的石墨化程度,如图8B 所示。观察到了D(1350 cm-1)和G(1580 cm-1)波段。其中,强度比(ID/IG)随着石墨化程度的增加而减小[32-33]。具体而言,75、150 和850 μm 的活性炭的ID/IG值分别为5.6、7.5 和3.9。这表明,850 μm的活性炭具有更高的石墨化程度,更低的电阻率和更优异的电催化性能。
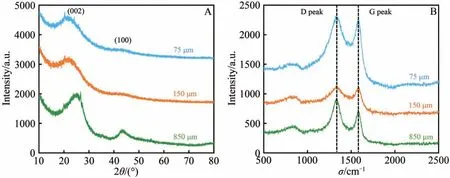
图8 不同粒径活性炭的XRD图(A)及拉曼光谱(B)Fig.8 XRD pattern (A) and Raman spectrum (B) of activated carbon with different particle sizes
利用XPS对不同粒径的活性炭进行了分析,结果如图9所示。通过去卷积N1s峰的分析,发现活性炭中主要存在石墨态N,而石墨态N 能够提高导电性并促进ORR 反应的进行[34-35]。这与拉曼光谱和RRDE 测试结果相一致。此外,图9F 显示了C—N 官能团百分比与ORR 电子转移数之间的高度线性关系,这表明C—N官能团的含量在H2O2生成过程中起着重要作用。然后,通过XPS对不同粒径活性炭所含的化学键类型及比例进行了分析,结果如图9D所示。不同粒径活性炭的C—N官能团比例存在明显差异,其中850 μm 的活性炭C—N官能团比例为39.3%。这进一步证明了850 μm的活性炭具有高选择性生成H2O2的能力。综上所述,850 μm 的活性炭不仅促进了ORR 反应的进行,而且其高C—N 官能团比例增强了二电子ORR反应的选择性,从而促进了AC@Ti-F体系中H2O2的生成。

图9 不同粒径活性炭的C1s分峰拟合图(A-C)、化学键占比(D)、N1s峰(E)及C—N占比与电子转移数相关性拟合(F)Fig. 9 C1s peak fitting diagrams (A-C),chemical bond proportion (D),N1s peak (E) and correlation fitting between C—N proportion and electron transfer number (F) of activated carbon with different particle sizes
2.4 AC@Ti-F GDE体系降解布洛芬效能
2.4.1 电过臭氧体系影响条件优化
为了更好地应用AC@Ti-F GDE 电过臭氧反应体系于实际废水处理,进一步研究了不同pH 值、电流和O3,gas浓度对布洛芬降解能力的影响,如图10所示。在不同pH值条件下,布洛芬的降解率在碱性条件下最低,在中性和酸性条件下趋势相似,但中性条件略优于酸性(图10A)。这可能是由于碱性条件下缺乏质子并且流动性较差[36-37],而酸性条件下O3的传质受到阻碍,同时在pH=3时,布洛芬主要以阳离子形式存在,而阴离子形式更有利于O3与布洛芬的反应[4],从而导致中性条件下反应最优。随着电流的增加,布洛芬的降解率也增加(图10B)。图10C的结果显示,布洛芬降解速率随着臭氧浓度的增加而增加,这是因为臭氧浓度的提高有助于促进O3从气相到液相的传质过程,进而促进O3与H2O2的反应以及在阴极还原过程中生成HO•等活性物质,从而增强布洛芬的降解[38]。综合考虑,AC@Ti-F GDE 体系的最佳运行条件为pH值为7,电流为150 mA,O3气体浓度为52.2 mg/L。然而,考虑到实际水厂处理污染物时通常采用0.5~1.0 mg(O3)/mg 溶解性有机碳(DOC)的浓度[39],因此后续研究将继续采用16.5 mg/L 作为实验运行条件。

图10 不同pH值(A)、电流(B)和臭氧浓度下(C)布洛芬降解情况Fig.10 Degradation of ibuprofen at different pH (A),current (B) and ozone concentrations (C)
2.4.2 降解机理
首先,通过ESR确定了AC@Ti-F GDE 体系中的自由基种类。其中,分别采用DMPO和TEMPO 作为捕获剂,以测定HO•和单线态氧(1O2)。而超氧阴离子自由基的测定则需要在甲醇溶液中使用DMPO作为捕获剂。实验结果如图11所示,可见AC@Ti-F GDE体系中存在和1O2等活性物质起到了降解和矿化污染物的作用。
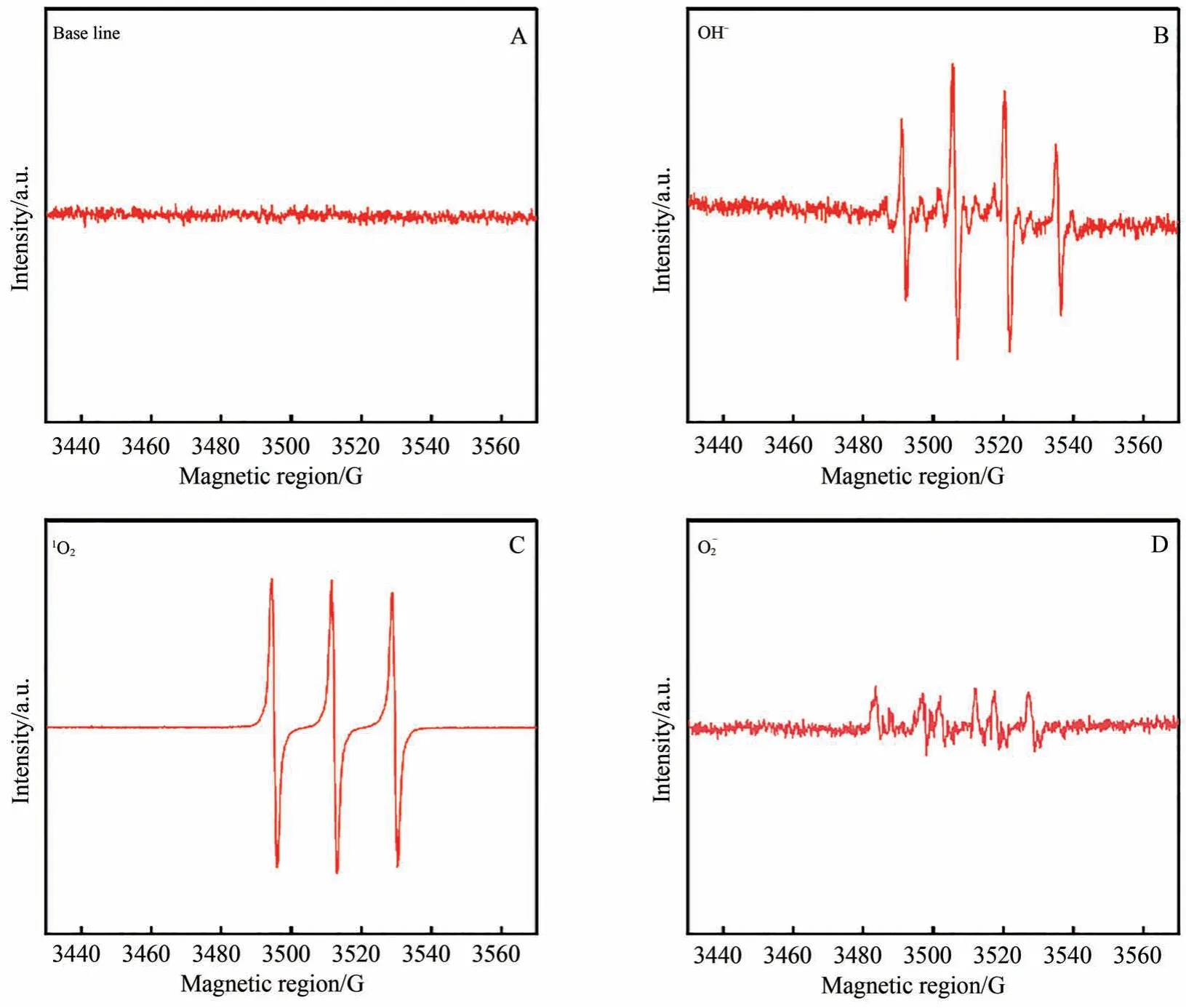
图11 空白样(A)、HO•(B)、1O2(C)和(D)的特征图谱Fig.11 Characteristic spectra of blank samples (A),HO• (B),1O2 (C) and (D)
在此基础上向体系中分别加入甲醇、糠醇和对苯醌等猝灭剂和过氧化氢酶,先与目标活性物质反应,探究不同活性物质降解布洛芬贡献程度。各种猝灭剂投加浓度为30 mmol/L,过氧化氢酶投加浓度≥500 U/mL。此外,为了研究吸附和阳极氧化对目标污染物去除效果的贡献程度,在与电过臭氧相同的条件下,未向水中添加任何猝灭剂,以探究阳极氧化过程的贡献程度;在与电过臭氧相同的条件下,不向水中添加猝灭剂和臭氧,进行吸附实验,探究吸附过程的贡献程度。图12A 显示了在反应前3 min内快速降解布洛芬,但后续受布洛芬传质受阻降解速度减慢,加入过氧化氢酶、甲醇、糠醇和对苯醌的降解体系布洛芬去除率依次降低。根据甲醇可以猝灭HO•,糠醇可以猝灭HO•和1O2,而对苯醌可以猝灭HO•和,然后通过图12A计算获得图12B,得到了不同降解路径对布洛芬的降解贡献。因此,AC@Ti-F GDE电过臭氧体系降解布洛芬阳极氧化贡献程度最大,其中和1O2,能大幅度提高布洛芬降解效能和降解速率,主要通过多种反应生成HO•短时间内快速去除目标污染物。

图12 投加不同猝灭剂后布洛芬降解情况(A)和降解贡献占比(B)Fig.12 Degradation of ibuprofen after adding different quenching agents (A) and degradation contribution ratio (B)
为确定AC@Ti-F GDE电过臭氧体系矿化布洛芬过程,测定了反应过程TOC变化情况(图13)。TOC随时间变化呈现先快速降低后缓慢降低的趋势,这可能是降解过程中不利于矿化的中间产物逐渐累积,影响体系中TOC进一步去除,在反应60 min后,TOC的去除率达到了52.3%。

图13 布洛芬矿化过程TOC变化情况Fig.13 Changes in TOC during the degradation of ibuprofen
随后,使用HPLC 和LC-MS 对布洛芬降解过程中的中间产物进行了检测,结果如表2 所示。表2 中A为布洛芬,B、C1和C2由布洛芬羟基化得到,随后发生去甲基化和支链氧化得到D、E、F、G和H,I是脱羧反应生成的芳香烃,J、K1和K2由支链进一步氧化得到,L、M、N1、N2、O、P和Q是芳香环开环生成的脂肪酸。

表2 LC-MS对布洛芬降解中间产物检测结果Table 2 Determination of ibuprofen degradation intermediates by LC-MS

Continued from previous page
结合已有的研究[21,42],推导出了AC@Ti-F GDE 电过臭氧体系中可能存在的布洛芬降解路径(图14)。在AC@Ti-F GDE 电过臭氧体系中,布洛芬首先发生羟基化反应,然后经过去甲基化和脱羧反应,使支链氧化并出现醛基和羰基等官能团,同时生成简单的芳香烃,随着反应的进行,HO•等活性物质进一步氧化支链,随后攻击苯环并开环生成脂肪酸,最终被矿化生成CO2和H2O。
最后,研究了AC@Ti-F GDE 在电解过程中的稳定性,结果如图15 所示。从电流-时间模式运行图中可以看出,每隔1 h 电流的波动是由于电解过程中多孔电极表面生成的小气泡会聚成大气泡后脱离电极。总体而言,在7 h的运行时间内,AC@Ti-F GDE体系仍然保持着稳定的电催化性质[43]。此外,经过10 次电解实验后,AC@Ti-F GDE 中的活性炭孔结构与反应前相比没有发生明显的改变(图15B 和15C)。因此,AC@Ti-F GDE具有出色的电催化稳定性,有潜力应用于实际废水处理过程中。

图15 电流-时间模式运行7 h情况(A);反应前(B)及电解10次后(C)电极SEM图Fig. 15 Current time mode operation for 7 h (A);SEM images of electrode before reaction (B) and after 10 electrolysis cycles (C)
3 结 论
通过在泡沫钛空气扩散电极内填充活性炭构建AC@Ti-F GDE 体系,对电过臭氧体系进行了强化研究。研究结果表明,相比于Ti-F、Ti-F GDE 和AC+Ti-F GDE 体系,AC@Ti-F GDE 能够通过建立气、固、液三相界面和微气泡曝气方式促进O2传质。填充的活性炭能够提高二电子ORR 反应的活性和选择性,从而强化H2O2的积累。同时,采用微气泡曝气方式可以促进O3传质,并催化H2O2与O3发生过臭反应,生成大量的HO•。
活性炭的投加密度可以通过增加活性炭催化O3的活性位点数量和影响O3传质来影响AC@Ti-F GDE 体系中HO•的相对含量。此外,活性炭的投加密度还可以通过影响活性炭与阴极金属接触形成微电场,从而影响AC@Ti-F GDE 体系中H2O2的积累。不同颗粒粒径的活性炭由于传质阻力和孔径分布的差异,会影响O3分子在活性炭中的停留时间,进而影响O3的催化反应。此外,活性炭的颗粒粒径还会对H2O2的催化分解和生成作用有影响,进而影响H2O2的积累。研究得出AC@Ti-F GDE 体系中最佳的活性炭投加密度为0.8 mg/cm3,最佳的颗粒粒径为850 μm。
将AC@Ti-F GDE 体系应用于降解布洛芬,研究得到最佳的运行条件为pH 值为7,电流为150 mA,质量浓度为52.2 mg/L。对布洛芬的降解机理和矿化过程进行了解析,并且阴极稳定性测试结果良好。这为将AC@Ti-F GDE体系应用于布洛芬废水预处理提供了理论依据。
猜你喜欢
煤气与热力(2021年10期)2021-12-02 05:11:46
童话世界(2020年32期)2020-12-25 02:59:18
中学生数理化·八年级物理人教版(2019年3期)2019-04-25 06:21:00
中学生数理化·八年级物理人教版(2017年12期)2017-04-18 12:59:46
中国蔬菜(2016年8期)2017-01-15 14:23:43
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:30
兽医导刊(2016年6期)2016-05-17 03:50:46
中国癌症杂志(2015年4期)2015-12-09 03:15:54
少儿科学周刊·少年版(2015年1期)2015-07-07 21:57:30
应用化工(2014年1期)2014-08-16 13:34:08
