
泼尼松合成方法综述
2024-03-15 02:27于旭超刘风强雷伟林建东
浙江化工 2024年2期
于旭超, 刘风强, 雷伟, 林建东
(台州仙琚药业有限公司, 浙江 台州 317016)
泼尼松(Prednisone, 1)又称强的松、去氢可的松,化学名称为:17α,21-二羟基孕甾-1,4-二烯-3,11,20-三酮(图1),具有抗过敏作用,能抑制结缔组织的增生, 属于肾上腺皮质激素类药物,具有抗炎及抗细血管壁和细胞膜的通透性,减少炎性渗出,并能抑制组胺及其他毒性物质的形成与释放。 泼尼松是泼尼松龙的前药,口服给药后经体内肝脏11β-羟基类固醇脱氢酶(11β-HSD)代谢转化为活性物泼尼松龙,临床上可用于系统性红斑狼疮、 药物性皮炎及自身免疫性疾病等治疗。 泼尼松水钠潴留及排钾作用比可的松小,抗炎及抗过敏作用较强,副作用较少,故临床上应用较为广泛[1-5]。
由于泼尼松为甾体类化合物, 合成路线长、工艺繁琐,实现工业化生产难度较大,因此工业制备泼尼松通常采用以甾体化合物为原料的半合成法。 因泼尼松独特的药理活性,其原料药的制备方法得到了医药工业领域的广泛重视,发展高效、绿色的泼尼松合成方法具有现实意义。 本文综述泼尼松合成方法,并对泼尼松合成工艺改进方向进行展望。
1 泼尼松的合成方法
1.1 合成路线1——以16,17-环氧黄体酮为原料
泼尼松的传统合成方法:以16,17-环氧黄体酮为原料,经微生物发酵、环氧开环、氢化、碘代、脱氢、水解等8 步反应制备(Scheme 1)[6-11]。 1950年,Petersen 等[6-8]通过采用霉菌发酵实现在孕酮第11 位引入羟基,这一关键发现为从黄体酮合成皮质类固醇提供了理论依据。以16,17-环氧黄体酮(2)为原料,用黑色根霉发酵引入11 位羟基,现已被广泛用于工业化生产。 虽然发酵单次转化率只有50%左右, 但由于反应选择性较好, 而且16,17-环氧黄体酮(2)与羟基化物3 易通过结晶分离, 因此原料2 可从母液中回收重复利用,使发酵过程的综合产率高于88%。11 位醇羟基经三氧化铬氧化可得到氧化物4, 进一步采用氢溴酸对16 位环氧开环,可得到溴羟化合物5,再经雷尼镍催化氢化还原得到化合物6。
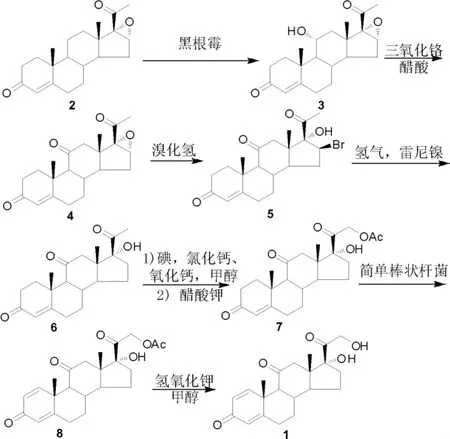
Scheme 1
随后,Stork 等[9]和Joly 等[10]对21 位甲基进行后续转化,经羰基α 位碘代及醋酸钾亲核取代得到醋酸酯中间体7。 目前工业生产中脱氢反应主要采用微生物发酵法,以棒状杆菌和诺卡氏菌为菌种, 以较高的选择性实现1,2 位脱氢化合物8的合成[11-12]。 在生物发酵法技术应用之前,化学法引入1,2 位双键曾被广泛使用。 其中,2,3-二氯-5,6-二氰基苯醌(DDQ)[13]和二氧化硒[14]是最常用的脱氢试剂。 然而,DDQ 用量大,反应液颜色深,后处理较为繁琐;二氧化硒的毒性较高,工业化应用受限。 因此,生物发酵脱氢法与化学脱氢法相比,成本、选择性等方面都更具优势。 最后将脱氢产物8 通过简单碱性条件下的水解反应得到泼尼松(1)。
此条工艺路线现已被广泛应用于泼尼松的工业化生产, 但该路线仍具有一定的缺陷:(1)11位醇羟基氧化需要依赖高价金属铬氧化,导致含铬废水量大,废液难处理;(2)生物发酵脱氢反应虽然已实现产业化,但现有发酵技术扔存在母液有效物含量少、发酵效率低、提取成本较高等问题。
1.1.1 氧化新方法
现有的合成工艺中,11 位羟基氧化通常采用三氧化铬,导致含铬废水量大,废液难以处理。 由于Cr6+具有较强的氧化性, 甾环其他位置上的双键、羟基耐受性不佳,导致氧化反应选择性差、收率低、杂质多。 研究人员已经开发部分11 位羟基氧化的改进方法。例如,卢彦昌等[15]提出了一种新的氧化11 位羟基的方法, 采用次氯酸钠为氧化剂,2,2,6,6-四甲基哌啶-N-氧化物(TEMPO)作为催化剂,溴化钾为助催化剂,室温条件下将11 位羟基氧化得到化合物6(Scheme 2),后续再经过碘代、亲核取代和水解反应,可制备得到泼尼松。该方法避免了重金属铬试剂的使用,反应条件更温和、绿色,易于工业化实施。

Scheme 2
此外,陈伟等[16]发展了一种11 位醇羟基的光催化氧化方法,以半导体材料二氧化钛为光催化剂,硝酸银为助催化剂,在深紫外光的照射下,激发光催化剂产生电子空穴,通过单电子转移过程将11 位的羟基氧化为羰基(Scheme 3)。上述方法采用绿色的氧气作为氧化剂,光敏剂及助催化剂均只需催化量,从工艺源头避免了化学当量强氧化剂的使用,工艺成本低,后处理简单。

Scheme 3
1.1.2 脱氢反应
近年来,通过化学法实现A 环脱氢反应的研究也获得了一定进展。例如,Olivetq 等[17]采用溴单质对原料二氢可的松-21-醋酸酯(11)进行1,4-位二溴代, 得到二氢可的松2,4-二溴代衍生物12,再经过3,5-二甲基吡啶脱溴化氢,最后在碳酸氢钾中水解得到泼尼松(1)(Scheme 4)。

Scheme 4
微生物发酵法是泼尼松A 环引入双键的常用方法,常见的发酵菌为棒状杆菌和诺卡氏菌[18]。后续的研究过程中发现,上述菌种在发酵反应过程中,除了能够实现脱氢反应外,也能够催化其他官能团转化(如碳碳双键、碳氧双键还原,羟基的氧化等), 也广泛应用于甾体侧链的分解或甾体环的降解过程。 研究表明,红球菌属生物除了能够实现各种固醇的羟基化或氧化外[19],也可以应用于甾环的脱氢反应,且比棒状杆菌和诺卡氏菌具有更高的选择性。 Costa 等[20]对13 株红球菌进行筛选,发现一种新型红球菌,以可的松为原料,可实现高效催化脱氢得到泼尼松(Scheme 5),摩尔收率可达94%。
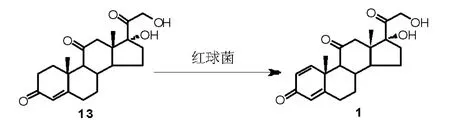
Scheme 5
1.2 合成路线2——以11α-羟基雄烷-1,4-二烯-3,17-二酮为原料
采用来源广泛、成本低廉的甾体化合物进行结构修饰是合成甾体药物的主要途径之一,例如雄甾酮类化合物已被大量应用于合成各种甾体药物,其D 环侧链经结构修饰可得到泼尼松类似物。 Marcel 等[21]以雄甾酮化合物14为原料,首先通过与金属锂试剂的缩合反应构建烯醚结构15,随后在对甲苯磺酸中加热搅拌,经烯醚水解后再经硼氢化钠甲醇溶液还原得到二醇物16。以间氯过氧苯甲酸(m-CPBA)作为环氧化试剂进行侧链烯烃的环氧化, 环氧化物中间体性质不稳定,很容易发生分子内环化/开环串联反应得到缩酮中间体17。 最后,缩酮17在酸性条件下水解得到化合物18(Scheme 6)。 该方法以廉价易得的雄甾酮类化合物为原料,为泼尼松侧链构建提供了新的思路,但使用的金属锂试剂用量较大,成本较高,其侧链构建方法不适合工业化生产。
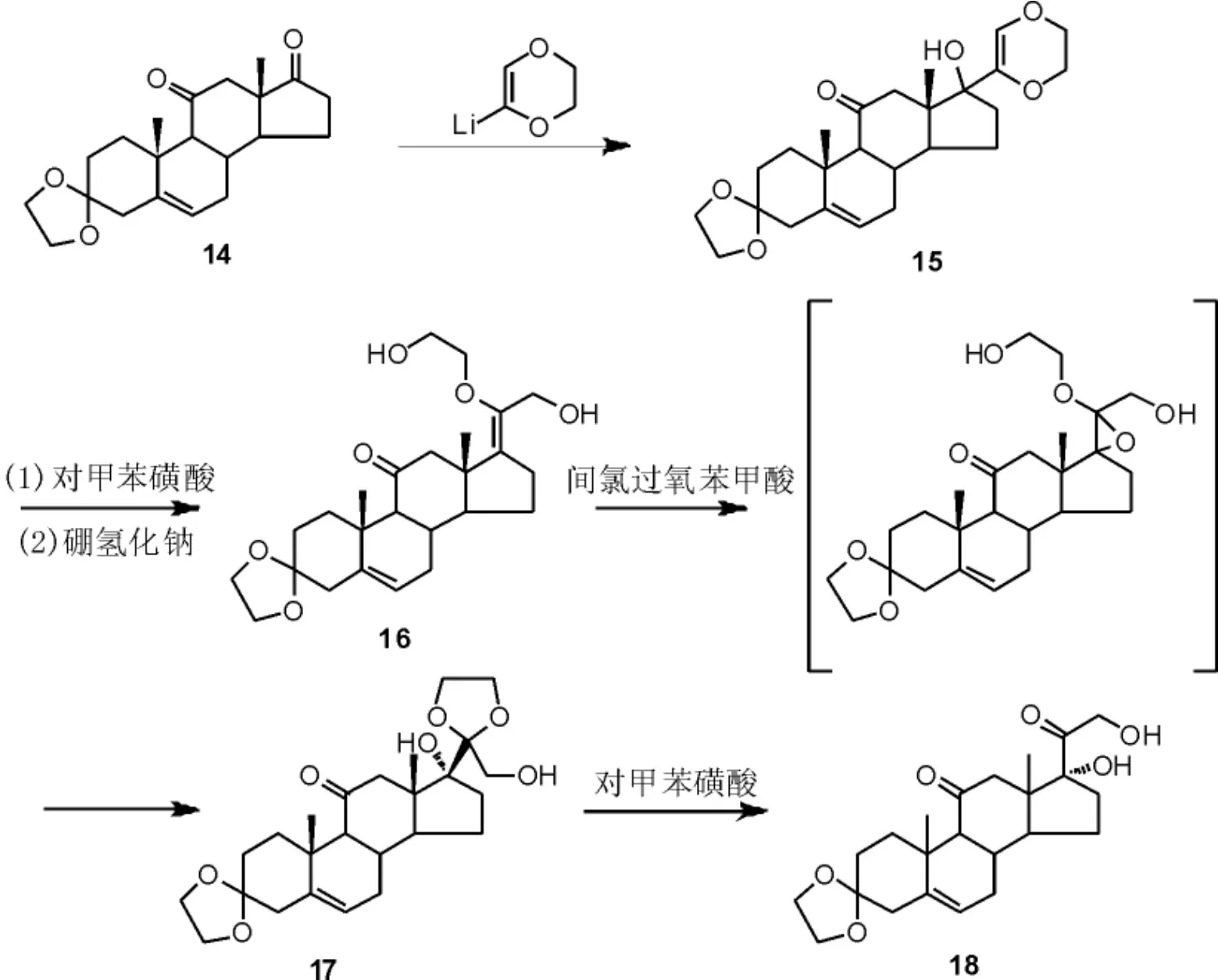
Scheme 6
刘喜荣等[22]报道了一条以11α-羟基雄烷-1,4-二烯-3,17-二酮(19)为原料合成泼尼松的路线。 首先,19经醋酸/三氧化铬体系氧化得到氧化物20。 随后,17 位羰基经丙酮氰醇氰基加成后,再对17 位羟基进行硅醚化得到中间体22。 在强碱正丁基锂中,22经分子内的亲核取代反应得到23, 再经乙酰氧基取代可得醋酸泼尼松(8)(Scheme 7)。 该路线提供了一种新的D 环侧链修饰方法,可避免传统路线中的碘代工艺,不使用毒性大、成本高的碘单质。 然而该路线需采用含氰试剂,导致含氰废水量大,工业化实施难度较大。
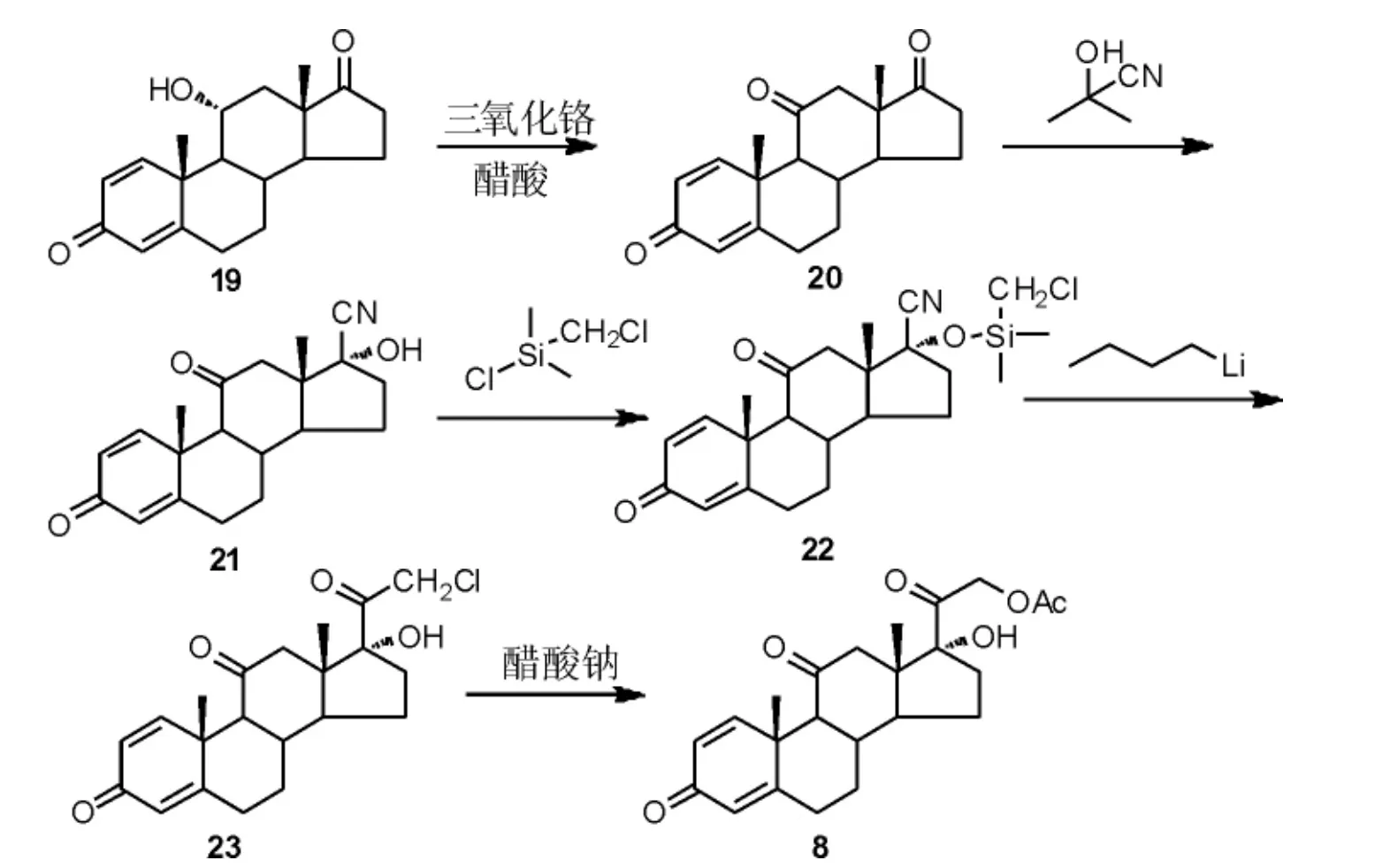
Scheme 7
1.3 合成路线3——以RSA 为原料
吴亚克等[23]以孕甾-4-烯17α, 21-二醇-3,20-二酮-21-醋酸酯(RSA,24)为原料,依次经过发酵羟化反应、发酵脱氢反应、氧化反应和水解反应制得泼尼松(Scheme 8)。 该制备方法采用大宗化的RSA 为起始原料,无需经过碘代再置换的步骤,合成路线短,总收率较高,但该方法11 位羟基氧化仍然需采用三氧化铬作为氧化剂。
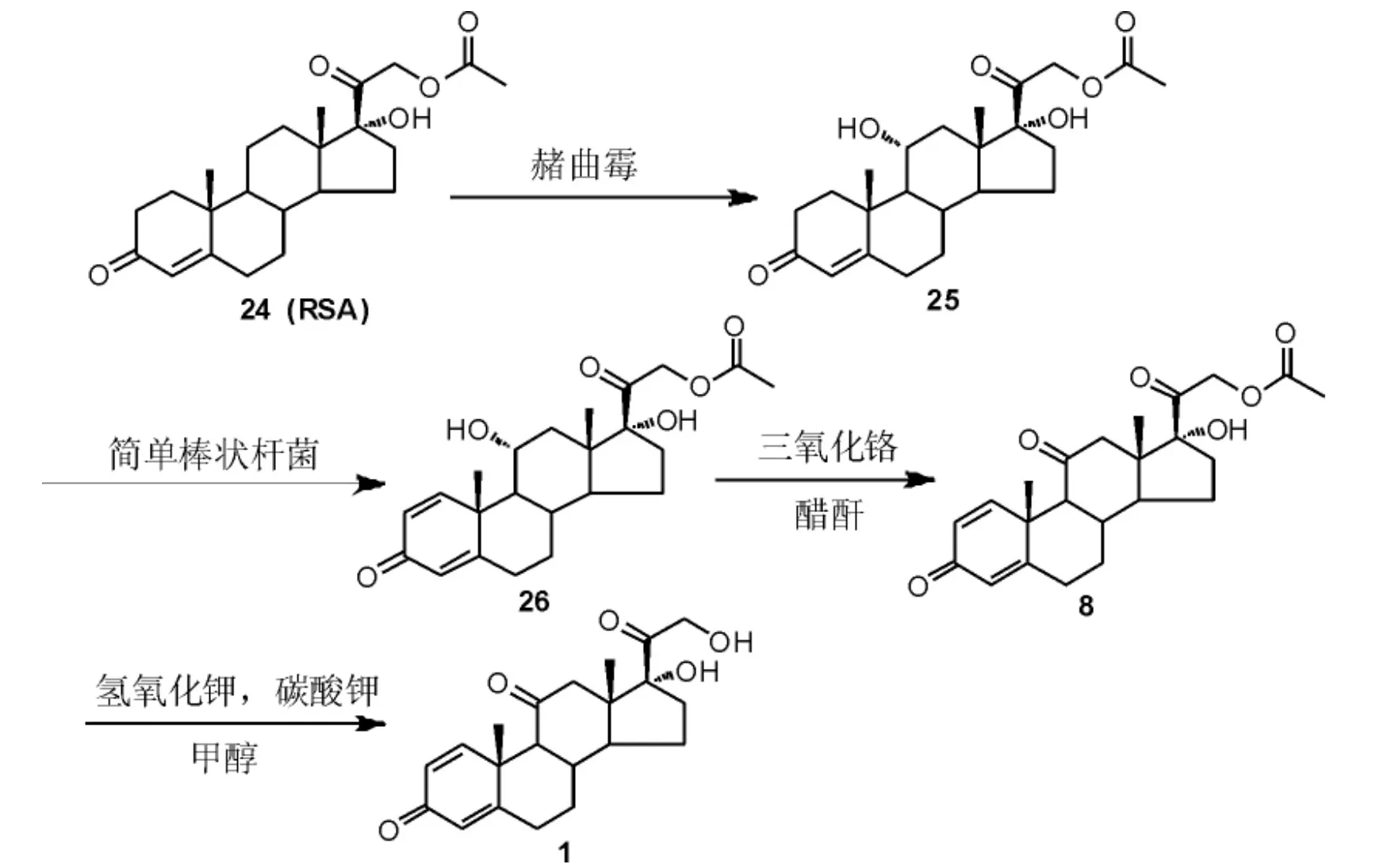
Scheme 8
1.4 合成路线4——以醋酸阿奈可他为原料
通常甾环化合物11 位羰基的引入需采用微生物发酵羟化、化学法氧化两步反应实现, Hicks等[24]报道一种通过双键亲电加成、氧化、还原脱溴等步骤引入11 位羰基的方法。 首先,在叔丁醇中添加适量硫酸,加入N-溴代乙酰胺,可得到溴羟化产物(28),实现了11 位羟基的引入(Scheme 9)。该方法提供了一种新的11 位羟基化方法, 再通过醇羟基氧化及C-Br 键还原即可制得目标化合物。 上述方法为甾体化合物的11 位羰基化提供了新的思路。
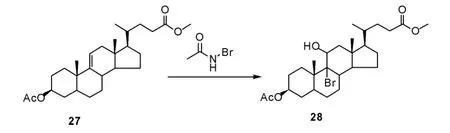
Scheme 9
王宇等[25]通过HClO4(高氯酸)/N-溴代丁二酰亚胺(NBS)体系实现了9 位双键的溴羟化。 在二醋酸碘苯(DIB)和碘单质存在下,通过光照得到氧化脱溴产物(Scheme 10)。两步反应产率仅有38%,而且反应过程中采用了毒性大、成本高的碘单质作为光反应引发剂。 因此发展更绿色温和的氧化/脱溴方法对于该工艺路线的工业化至关重要。
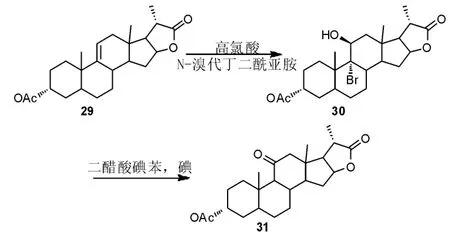
Scheme 10
陈伟等[26]以醋酸阿奈可他为原料,经溴羟化、脱溴重排两步反应引入11 位羰基(Scheme 11)。首先,在甲磺酸催化下,以二溴氰基乙酰胺为溴化剂,原料32发生溴羟化反应得到溴羟物33,再经过吡啶氢溴酸脱溴重排, 得到氧化产物7。 最后,化合物7经过简单诺卡氏菌和灰色链霉菌两种微生物发酵实现1,2 位脱氢, 并经水解得到最终产物泼尼松。 该工艺路线短,避免了三氧化铬、碘单质等有毒有害试剂的使用,更加绿色安全。

Scheme 11
2 总结及展望
(1)4 种泼尼松的合成路线都存在一定的缺陷, 前3 条路线均需采用三氧化铬实现11 位羟基的氧化,虽然研究人员已发展了几种氧化的替代方法,但相关研究由于生产成本高、放大重复性差等问题,暂无法满足工业化,主要停留在实验室小试阶段。 另外,路线1 中还存在路线长,氢气催化氢化存在安全隐患等问题;路线2 采用剧毒试剂丙酮氰醇作为氰源, 废水难以处理,且LDA 催化的分子内取代反应条件苛刻, 重复性差; 路线4 存在原料醋酸阿奈可他的来源少,成本较高的问题。
(2)随着泼尼松的市场需求越来越大,绿色、安全的制药工艺路线的开发更加重要。 未来泼尼松合成路线的研究方向主要包括: ①寻找来源广、更加容易修饰的天然甾体原料化合物,进一步缩短泼尼松的合成路线; ②改进现有11 位羟基氧化工艺,光催化氧化已在合成方法学上获得突破,随着新型光催化反应器的出现,光催化氧化反应将取得良好的放大效应,有望尽早实现对三氧化铬氧化工艺的替换;③随着合成生物学理论和技术的不断发展, 可以优选出更加专一、高效的菌种实现选择性脱氢、羟化反应,提高工艺的整体收率。
猜你喜欢
云南化工(2021年11期)2022-01-12
中华养生保健(2020年1期)2020-11-16
海洋通报(2020年2期)2020-09-04
广州化工(2020年8期)2020-05-19
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年5期)2018-06-13
中成药(2017年4期)2017-05-17
华东理工大学学报(自然科学版)(2015年5期)2015-02-27
中国药业(2014年24期)2014-05-26
中国医学科学院学报(2014年6期)2014-03-11
