
前驱体预处理对富锂锰基氧化物电化学的影响
2024-03-13 07:36文萌夏鼎峰钟盛文
有色金属科学与工程 2024年1期
文萌, 夏鼎峰, 钟盛文*
(江西理工大学,a.材料冶金化学学部;b.江西省动力电池及材料重点实验室,江西 赣州 341000)
近年来,由于碳中和与全球能源危机,可再生清洁能源已经引起了很多关注,例如太阳能、风能和水能。这些可再生能源急需具有稳定存储能力和高转换效率的储能装置。锂离子电池是一种常见的储能设备,具有高能量转换效率、高能量密度和长寿命性能,已经被应用于许多智能便携式设备中[1-6]。这些设备需要更高能量密度以满足人们需求。相比其他正极材料,富锂锰基材料因其较高的容量(>250 mAh/g),因此被视为最有潜力的下一代正极材料。然而,富锂锰基材料存在以下问题,例如:首次库仑效率低、电压衰减、倍率性能差,从而限制了其商业化进程[7-9]。
目前,研究人员主要通过对富锂锰基材料进行掺杂、表面改性和改变电压制度来缓解上述方面的问题,例如通过掺杂F-增加材料结构稳定性从而改善电压衰减。与此同时,也可以通过控制粒径大小来改善动力学迟缓和倍率性能差等问题[10]。此外,通过改变材料颗粒大小,可以改善动力学滞后和不良的倍率性能。前人在共沉淀合成过程中,通过调节pH值[11]、浓度和转速等参数合成了不同形态和尺寸的前驱体[12-16],以前的方法在合成过程中影响因素较多,难以保证每次合成过程中各参数的一致性。本实验提供了一种改变颗粒尺寸的方法,可以极大提高实验重复性。本文将共沉淀法合成的Ni0.3Mn0.7(OH)2前驱体用气流粉碎机进行机械粉碎,得到形状和粒径不同的2种前驱体。将干燥的前驱体和LiOH以n(M)∶n(Li)=1∶1.55(M=Ni+Mn)在卫星球混合器中混合。混合后的产品在900 ℃的炉子里以0.2 m3/s的空气气氛烧结14 h,得到2种不同粒度的阴极材料Li1.2Ni0.24Mn0.56O2(D50分别为1.667 µm和1.148 µm)。
1 实验方法
1.1 正极材料的制备
以六水硫酸镍(NiSO4·6H2O)、一水硫酸锰(MnSO4·H2O)、氢氧化钠(NaOH)和氨水(NH3·H2O)为原料在连续搅拌釜式反应器(CSTR)中合成Ni0.3Mn0.7(OH)2前驱体。过渡金属溶液的进料速率固定为 1.1 mL/min,基础溶液(NaOH 和 NH3·H2O的混合物)的泵送速率由pH=11控制。沉淀反应在恒温(55 ℃)和固定搅拌速度(450 r/min)下进行。将N2保护气体引入 CSTR 以防止 Mn2+氧化。搅拌反应8 h后,静置陈化2 h,而后过滤、水洗至中性,得到前驱体。在120 ℃的真空烘箱中干燥 24 h。将有无经过气流磨处理的前驱体Ni0.3Mn0.7(OH)2和 LiOH 以1∶1.55摩尔比混合均匀,后在550 ℃/6 h至900 ℃/14 h下进行高温烧结,制备出不同粒径大小的富锂锰正极材料 Li1.2Ni0.24Mn0.56O2。
1.2 材料结构与形貌表征
采用BT-9300ST 型激光粒度仪对所得到的材料粒径进行表征。采用日本理学MiniFlex 600型转靶衍射仪(XRD)对产物进行物相分析,测试条件为:Cu Kα辐射,λ=0.154 06 nm,60 kV管电压,50 mA管电流,扫描范围2θ=10°~80°,步长为0.02°,扫描速率为2 (°)/min;采用德国ZEⅠSS EVO/MA10 型扫描电镜表征对所得到的材料形貌进行表征。采用德国ZEⅠSS EVO/MA10 型BET对所得到的材料进行比表面积测试。采用X射线光电子能谱仪(XPS;PHⅠ5700 ESCA) 分析表面过渡金属元素(Mn、Ni)的氧化数。通用结构分析系统 (GSAS) 用于对所有化合物进行结构细化。
1.3 电化学性能测试
将富锂锰基正极材料Li1.2Ni0.24Mn0.56O2、黏结剂PVDF、导电剂SP 三者按质量比90∶6∶4称取,固体含量按45%(指质量分数)来确定NMP的量。将PVDF溶于NMP中直至无气泡时,再将称取好的正极材料、导电材料SP放入研钵中手动研磨均匀后,最后将溶解好的PVDF加入混合均匀的固体材料中,在匀浆机中高速转动以形成浆液。用这种浆料涂覆铝箔,并在 120 °C 下干燥 30 min。随后进行滚压冲片制备直径为12 mm的圆盘。将圆盘称重后在70 °C的真空温度下再干燥12 h。电解液的制备为碳酸亚乙酯(EC) 、碳酸甲乙酯(EMC)和碳酸二甲酯(DMC)的混合物(EC∶EMC∶DMC的体积比例为1∶1∶1),然后将电解质LiPF6(1 mol/L)溶解在该混合物中。隔膜由聚丙烯-聚乙烯-聚丙烯微孔薄膜(Cellgard 2400)制成。将硬币型电池(CR2032)组装在充满氩气的手套箱中,锂片作为对电极。电压设定为 2.0~4.6 V,使用中国深圳市新威尔电子有限公司生产的电化学测试仪设备进行电化学性能测试。所有电化学实验均在 25 °C 下进行。
2 结果与讨论
2.1 粒度分析
图1描述了材料的制备过程。Ni0.3Mn0.7(OH)2(D50=1.626 µm)样品的激光粒度图谱如图2(a)所示,Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)样品的激光粒度图谱如图2(b)所示。图2(a)显示了氢氧化物前驱体的粒度分布。大多数直方图曲线在0.5 µm和2 µm处具有最大值,分别为晶核、附聚体碎片。Ni0.3Mn0.7(OH)2(D50=0.710 µm)样品峰值强度在粒径为0.5 µm时远高于2 µm的沉淀物,表明沉淀物主要是以晶核的形式存在[17]。Ni0.3Mn0.7(OH)2(D50=0.710 µm)样品峰值强度在2 µm高于0.5 µm的沉淀物,表明沉淀物主要是以附聚体碎片的形式存在。与此同时,如图2(b)所示,Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和1.148 µm)样品的激光粒度生长符合正态分布。

图1 Ni0.3Mn0.7(OH)2的合成处理和Li1.2Ni0.24Mn0.56O2的合成过程流程示意Fig.1 Flow chart of the synthetic treatment of Ni0.3Mn0.7(OH)2 and the synthetic process of Li1.2Ni0.24Mn0.56O2
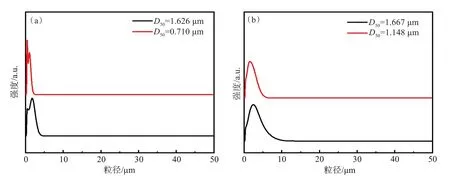
图2 不同粒径的前驱体及其对应粒径的正极料样品的激光粒度图谱:(a) Ni0.3Mn0.7(OH)2 (D50=1.626 µm)样品;(b) Li1.2Ni0.24Mn0.56O2 (D50= 1.148 µm)样品Fig.2 Laser particle size mapping of precursors of different particle sizes and their corresponding size of cathode material samples:(a) Ni0.3Mn0.7(OH)2 (D50=1.626 µm);(b)Li1.2Ni0.24Mn0.56O2 (D50= 1.148 µm)
2.2 前驱体材料结构分析
通过粉末XRD谱验证了Ni0.3Mn0.7(OH)2前驱体的晶体结构,如图3所示。图3中用心型符号突出显示的小峰是Mn2+氧化的结果。结果表明,即使在合成过程中通入了N2保护气体,仍然会再合成,洗涤或干燥过程中存在部分Mn2+氧化为Mn3+。位于2θ=19°的主峰,由于初级粒子的各向异性生长,强度比其他峰高得多。从图3中可以明显看出Ni0.3Mn0.7(OH)2(D50=0.710 µm)的主峰强度高于Ni0.3Mn0.7(OH)2(D50=1.626 µm),表明经过气流磨处理后的前驱体具有更高的各向异性生长。根据文献[7]的报道,35°~40°之间的宽峰与金属氢氧化物沿c轴的堆垛层错有关,该层错与β-Ni(OH)2相具有相等结构。
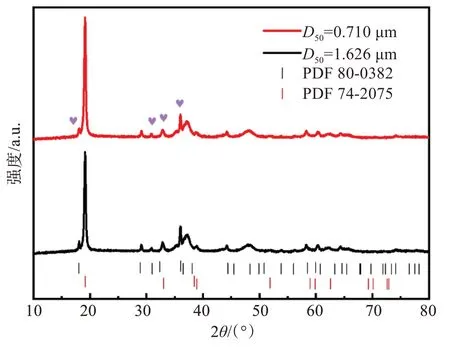
图3 Ni0.3Mn0.7(OH)2 (D50=1.626 µm和0.710 µm)样品的XRD图谱Fig.3 XRD patterns of Ni0.3Mn0.7(OH)2 (D50=1.626 µm and 0.710 µm) samples
2.3 前驱体材料SEM分析
为了确定经过气流磨处理前后的前驱体形貌和颗粒大小,进行了SEM测试。如图4(a)所示,未经气流磨处理的前驱体显示出一次片状组成的二次团聚体。如图4(b)所示,经过气流磨处理后的前驱体呈一次分散片状。

图4 前驱体样品扫描电镜图像:(a) Ni0.3Mn0.7(OH)2 (D50=1.626 µm)样品;(b) Ni0.3Mn0.7(OH)2 (D50=0.710 µm)样品Fig.4 Scanning electron microscope images of precursor samples:(a) Ni0.3Mn0.7(OH)2 (D50=1.626 µm);(b) Ni0.3Mn0.7(OH)2 (D50=0.710 µm)
2.4 正极材料结构分析
为了确定粒径大小对富锂锰材料结构的影响,进行XRD测试。如图5(a)所示,所有反射的主峰都指向六方晶系α-NaFeO2型层状结构,属于R-3m空间点阵群,还出现了Li2MnO3相的特征峰。与此同时,没有出现其他的杂峰,这说明合成的材料具有较高的纯度。由图5(b)可以看出颗粒粒径变小后 (003)峰向左偏移,表明材料层间距在增大。2个样品在(006)/(012)和(018)/(110)处均显示出明显的相邻峰分裂,表明形成了层状结构。各样品的c/a值均大于4.899,说明材料具有很好的层状结构。I(003)/I(104)可用于表征阳离子混排程度,I(003)/I(104)的值越高,材料中的阳离子混排程度越低。从表1中可以清晰地看出Li1.2Ni0.24Mn0.56O2(D50= 1.667 和 1.148 µm)2种材料样品的I(003)/I(104)比值均大于1.2,说明2种材料样品的阳离子混排程度较低。对比可以发现,Li1.2Ni0.24Mn0.56O2(D50=1.148 µm) 材料样品的I(003)/I(104)比值明显大于Li1.2Ni0.24Mn0.56O2(D50=1.667 µm)材料样品的I(003)/I(104)比值,说明Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)材料样品具有更低的阳离子混排程度[18-21]。
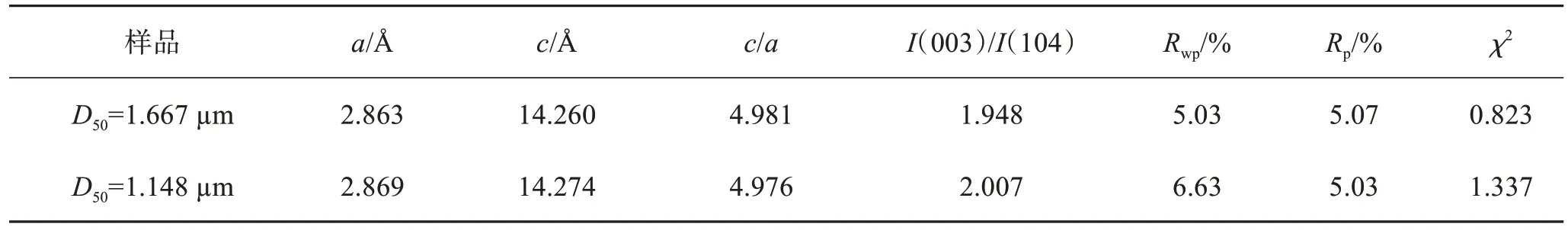
表1 Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和1.148 µm)样品R-3m和C2/m相晶格参数Table 1 R-3m and C2/m phase lattice parameters of Li1.2Ni0.24Mn0.56O2 (D50=1.667 and 1.148 µm) samples
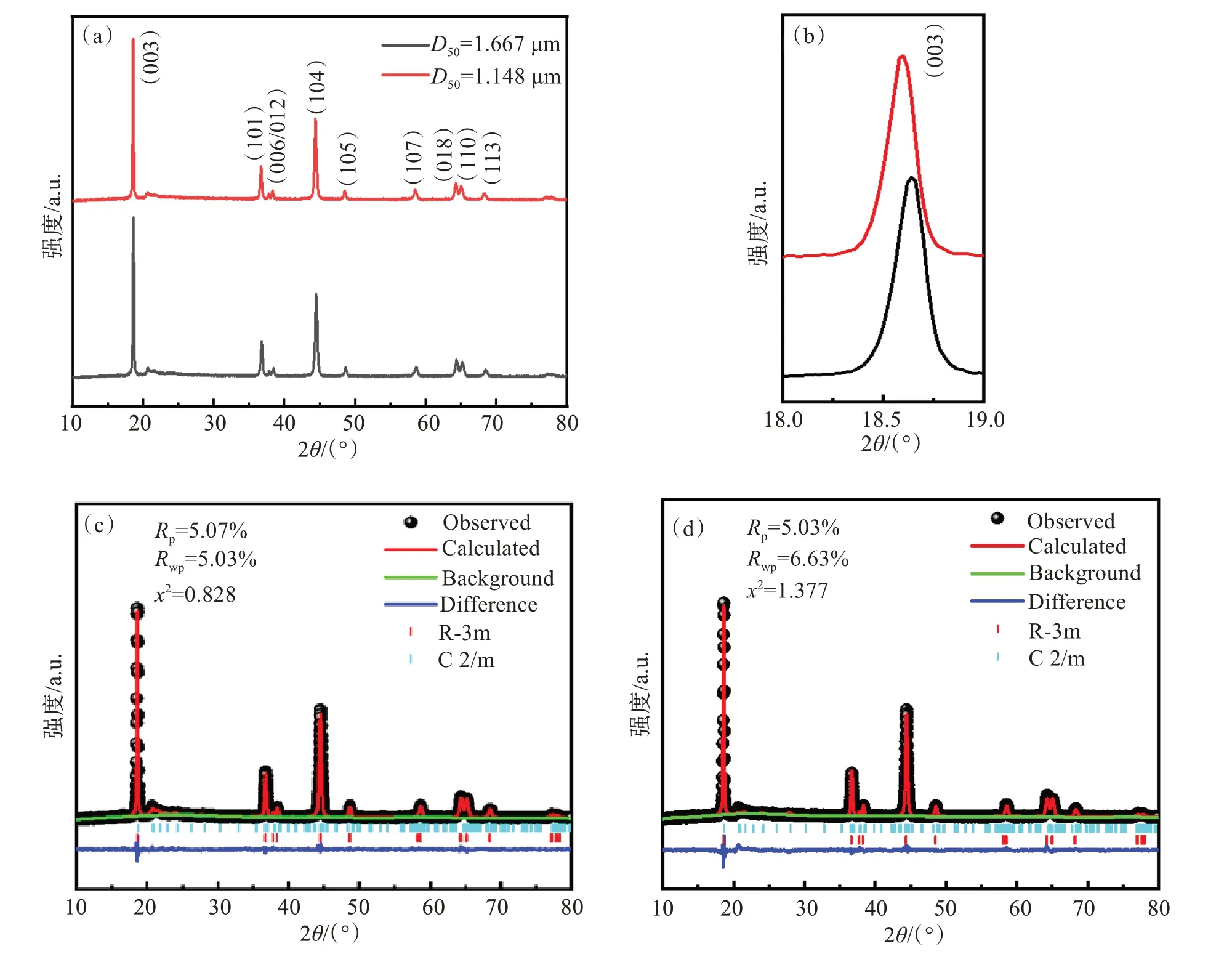
图5 Li1.2Ni0.24Mn0.56O(2D50=1.667 和 1.148 µm)样品的XRD图谱和XRD精修图谱:(a) XRD图谱;(b)( 003)峰的XRD放大图;(c) D50=1.667 µm样品的XRD精修图谱;(d) D50=1.148 µm样品的XRD精修图谱Fig.5 XRD patterns and XRD refined patterns of Li1.2Ni0.24Mn0.56O2( D50=1.667 µm and 1.148 µm) samples:(a)XRD pattern ;(b) the magniffed splitting pair peak of( 003);(c) riveted reffnement results of D50=1.667 µm sample ; ( d) riveted reffnement results of D50=1.148 µm sample
为了确定粒径大小对菱面体 LiMO2(R-3m)相和单斜Li2MnO3(C2/m)相组分含量的影响,通过GSAS软件进行XRD Rietveld研究了相组成,具体拟合数据见表1。
2.5 正极材料SEM和BET分析
为了确定不同粒径下正极材料的形貌和颗粒大小进行了SEM测试。结果显示,所有样品均由一次颗粒堆积成的二次颗粒组成,同时部分样品在视觉上具有锋利的边缘和光滑的小平面(图6(a)和图6(b),现象表明这些颗粒具有良好的结晶性。为了确定一次颗粒的大小。进行了粒径统计,如图6(c)和图6(d)对比显示Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)样品具有微弱的一次颗粒团聚和最小的一次颗粒平均粒径。Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和 1.148 µm)材料样品的BET测试结果如图7所示。Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和 1.148 µm)的BET比表面积分别为2.613 2 m2/g和3.017 3 m2/g,可以得知粒径变小后颗粒获得较大比表面积。
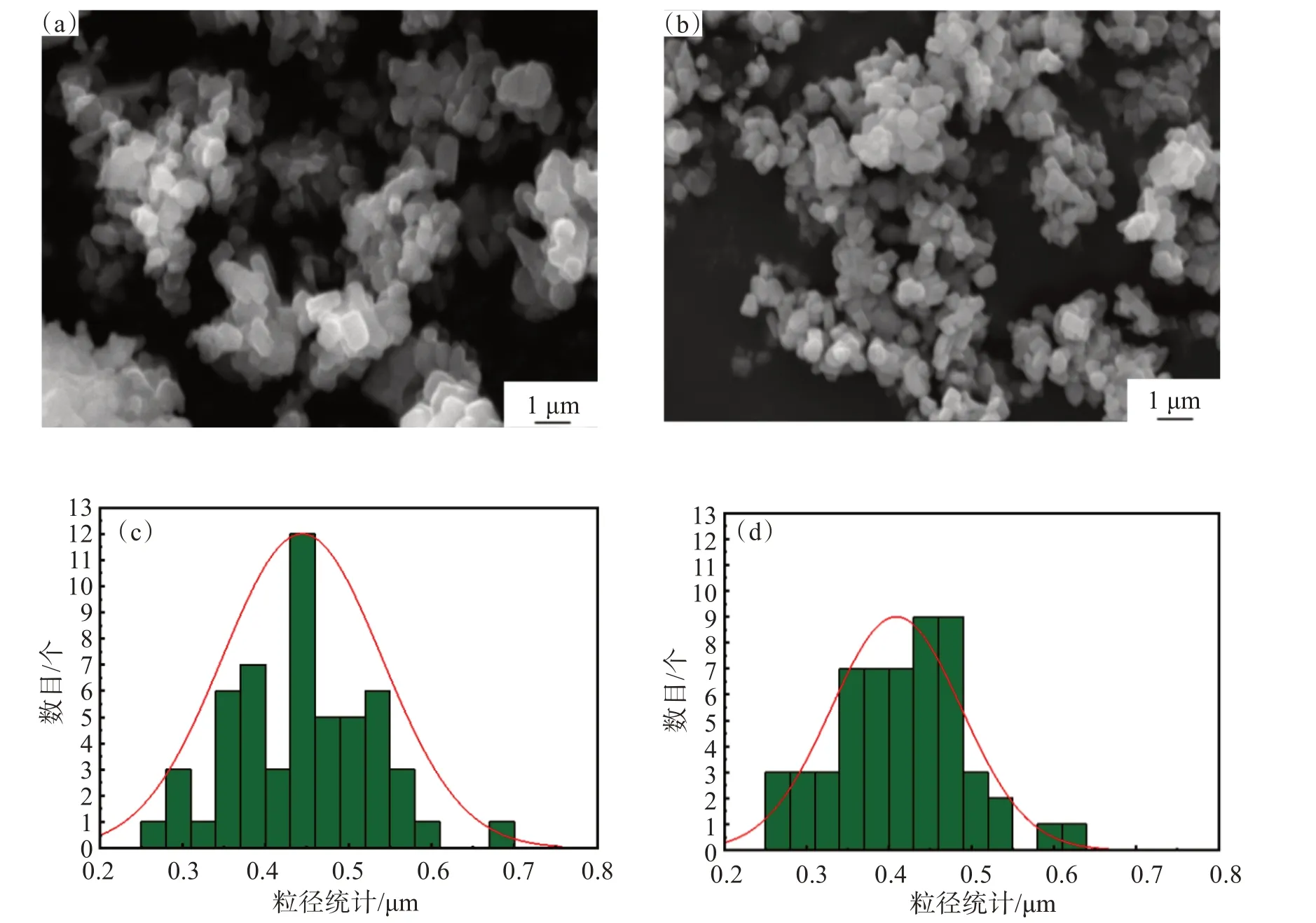
图6 Li1.2Ni0.24Mn0.56O2 样品扫描电镜图像和粒度统计图:(a) D50=1.667 µm样品的SEM图谱;(b) D50=1.148 µm样品的SEM图谱;(c) D50=1.667 µm样品的粒径统计图谱;(d) D50=1.148 µm样品的粒径统计图谱Fig.6 SEM images and particle size statistics of Li1.2Ni0.24Mn0.56O2 samples: (a) SEM image of D50=1.667 µm sample ; (b) SEM image of D50=1.148 µm sample (c) particle size statistics of D50=1.667 µm sample;(d) particle size statistics of D50=1.148 µm sample
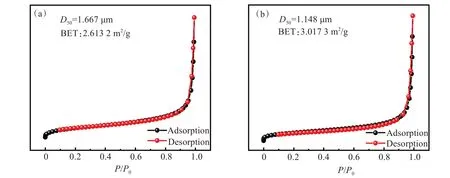
图7 Li1.2Ni0.24Mn0.56O2 样品的N2吸附-解吸等温线:(a) D50=1.667 µm样品的N2吸附-解吸等温线;(b) D50=1.148 µm样品的N2吸附-解吸等温线Fig.7 N2 adsorption-desorption isotherms of Li1.2Ni0.24Mn0.56O2 samples:(a) N2 adsorption/desorption isotherms for D50=1.667 µm sample; (b) N2 adsorption/desorption isotherms for D50=1.148 µm sample
2.6 正极材料XPS分析
为了确定过渡金属离子的价态,对2个样品进行了XPS分析。如图8所示,在2个样品的XPS测量中都观察到Ni、Mn、O、C和Li元素的信号。图8(b)和图8(c)分别显示了Mn 2p和Ni 2p的拟合XPS光谱,峰面积总结在表2中。Mn 2p的XPS光谱(图8(b))在642.5 eV的主峰与Mn4+的模式相符641.5 eV处的微弱峰值与Mn3+的模式很匹配。随着粒度的减小,Mn3+的峰面积从17.4%增加到18.5%(表2),而Mn4+的量从82.6%减少到81.5%。Ni 2p的XPS光谱(图8(c))显示,854.5 eV处的主峰与Ni2+匹配,856.2 eV处的主峰与Ni3+匹配。随着粒径的减小,Ni2+含量减少,锂镍混排降低[22-24]。

表2 XPS图中的Mn2+/Mn3+和Ni2+/Ni3+相关峰面积数据Table 2 Peak area data related to Mn2+/Mn3+ and Ni2+/Ni3+ in XPS plots
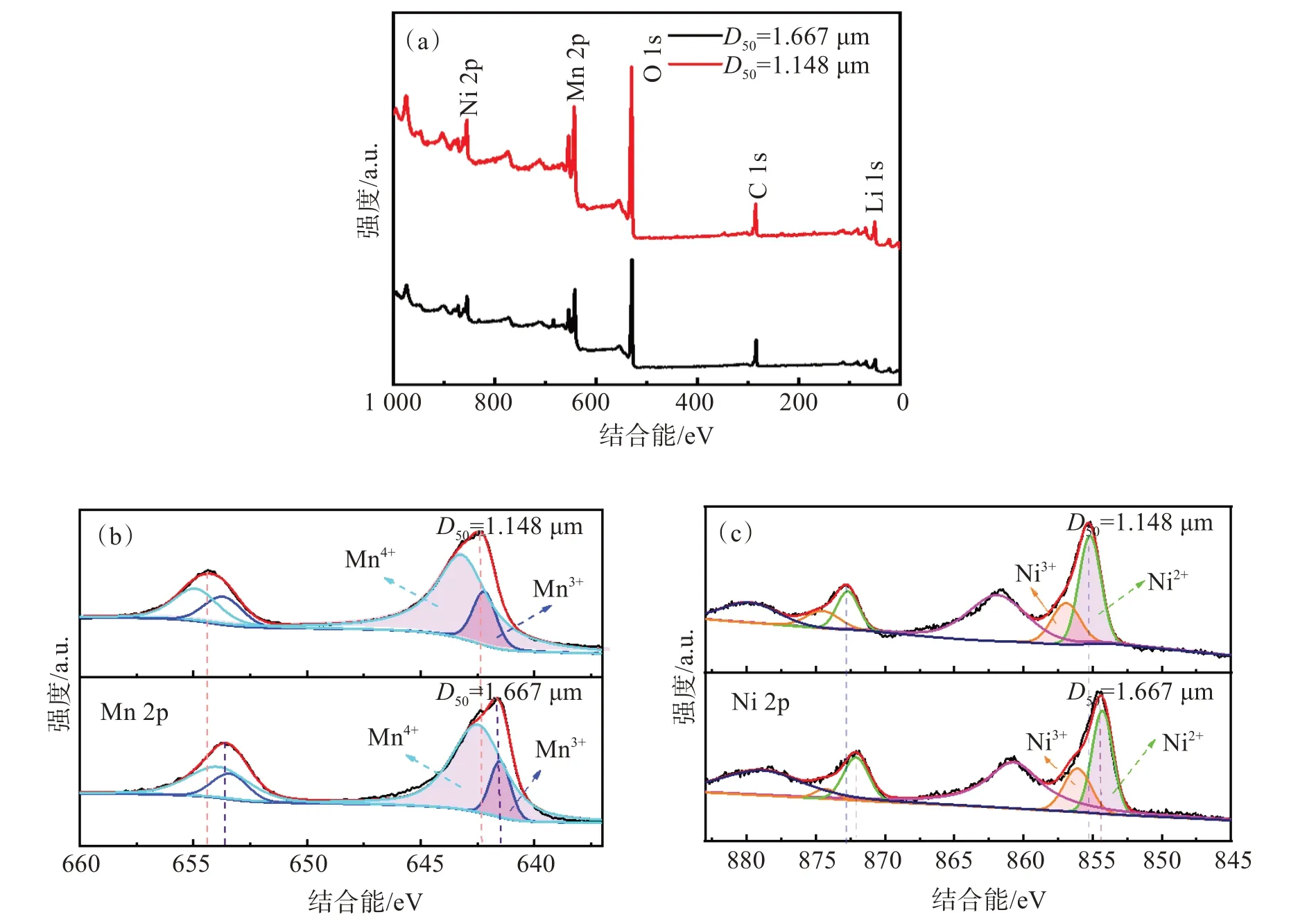
图8 所制备样品的XPS谱:(a) XPS总谱;(b) Mn 2p的XPS光谱;(c) Ni 2p的XPS光谱Fig.8 XPS spectrum of the prepared samples: (a)XPS spectra; (b) Mn 2p XPS spectra for sample;(c) Ni 2p XPS spectra for sample
2.7 电化学性能分析
Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和 1.148 µm)电极在2.0~4.6 V(C/10)的首次充放电曲线如图9(a)所示,工作电极的放电比容量和库仑效率具体数值如表3所示。可以清楚地看出,小粒径相对于大粒径的电极初始放电比容量和相应的库仑效率均增大[25-27]。

表3 Li1.2Ni0.24Mn0.56O2(D50=1.667 µm和1.148 µm)的初始充放电数据Table 3 Initial charge-discharge data of Li1.2Ni0.24Mn0.56O2 (D50 = 1.667 µm and 1.148 µm)
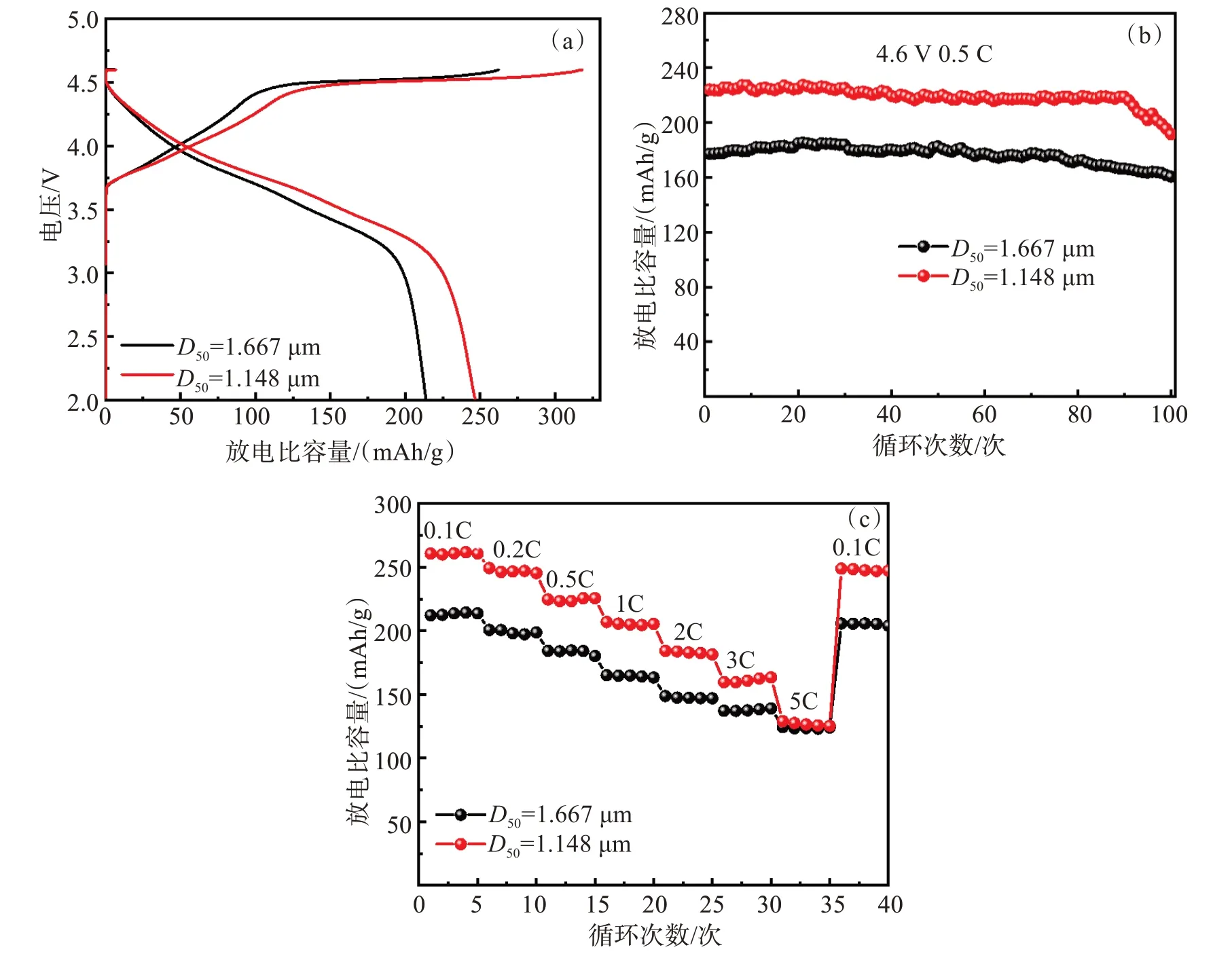
图9 Li1.2Ni0.24Mn0.56O2 (D50=1.667 µm和 1.148 µm)的电化学性能曲线:(a) 首次化成曲线;(b) 0.5 C循环曲线;(c) 倍率曲线Fig.9 Electrochemical performance curves of Li1.2Ni0.24Mn0.56O2 (D50=1.667 µm and 1.148 µm): (a) initial charge-discharge curves ;(b)cycling stability at 0.5C;(c) the rate capability curves
如图9(b)所示,在2.0~4.6 V范围内 0.5 C电压下充电-放电循环曲线显示,含Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)正极的比容量在循环过程中始终高于Li1.2Ni0.24Mn0.56O2(D50=1.667 µm)。在100次循环后,Li1.2Ni0.24Mn0.56O2(D50=1.667和1.148 µm)剩余比容量值分别为190.7 mAh/g和 237.3 mAh/g,相应的循环保持率为91.2%和90.9%。循环稳定性降低是由于粒径变小后比表面积大幅增加,反应活性随之提高,引起颗粒与电极液之间副反应增多[1-3]。如图9(c)所示,Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)电极相比Li1.2Ni0.24Mn0.56O2(D50=1.667 µm)表现出更高的倍率性能。主要原因是由于粒径变小大大缩短了锂离子在材料内部传输扩散的路径,有利于锂离子在材料晶格内部的快速脱嵌;同时粒径变小具有更大的比表面积,增大了电解液与材料的接触面积,表明锂离子脱嵌的活性位点增多,增大了倍率性能。
3 结 论
1)综上所述,通过前驱体预处理合成了不同颗粒大小的富锂锰正极材料。Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)材料样品显示出最小的平均粒径和最大比表面积。
2)电化学测试结果表明,颗粒大小对富锂层状氧化物的电化学性能具有重要意义。这种独特的形态产生了优异电化学动力学,这使得Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)材料样品具有较优的电化学性能。Li1.2Ni0.24Mn0.56O2(D50=1.148 µm)正极材料在0.1 C时的初始放电比容量为246.7 mAh/g。在0.5 C循环100次后,容量为190.7 mAh/g,循环保持率为91.2%。
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
南京大学学报(自然科学版)(2021年1期)2021-01-30
表面工程与再制造(2019年6期)2019-08-24
资源节约与环保(2018年1期)2018-02-08
池州学院学报(2017年3期)2017-10-16
材料科学与工程学报(2016年1期)2017-01-15
系统工程与电子技术(2016年12期)2016-12-24
当代化工研究(2016年7期)2016-03-20
电源技术(2015年9期)2015-06-05
