
超高效液相色谱-串联质谱检测枸杞中苦参碱和氧化苦参碱残留量的方法
2024-03-13 12:27:26张闯赵孔祥潘红蕊于艳军宓捷波苏明跃杨永超
食品研究与开发 2024年5期
张闯,赵孔祥,潘红蕊,于艳军,宓捷波,苏明跃,杨永超
(1.天津海关工业产品安全技术中心,天津 300457;2.天津海关动植物食品检测中心,天津 300457)
苦参碱农药是从苦参属植物中分离出来的全部物质,其主要成分为苦参碱和氧化苦参碱[1]。苦参碱与氧化苦参碱作为杀虫剂被广泛应用于国内外农业生产[2]。目前苦参碱农药是我国植物源农药中登记企业最多的品种,目标害虫有蚜虫[3],红火蚁[4-5]、金龟子甲虫[6]、田间幼虫[7]、黑腹果蝇、美洲大蠊[8]等,应用于豇豆生产种植[9]、结球甘蓝防控[10]、茶叶种植防治[11]等领域。其作用机理是害虫触及苦参碱农药后被麻痹神经中枢,苦参碱农药作为杀虫剂可凝固虫体蛋白质,阻塞气孔,使害虫窒息而死,起到触杀和胃毒的双重作用[12-13]。由于其杀虫活性相对较低,多与其他杀虫剂配伍使用[14]。相关研究表明,苦参碱农药对人体有较为明显的副作用。例如氧化苦参碱的毒性能降低人体正常肝细胞活性,大幅提高乳酸脱氢酶水平,通过光镜观察,人体肝细胞缩小、变得褶皱,显示出肝毒作用[15]。
枸杞是我国药食同源的中药材重要品种,因其茎叶繁茂、果汁甘甜、营养丰富,易遭受病虫的危害[16]。苦参碱、氧化苦参碱作为枸杞主要杀虫剂,可防治枸杞蚜虫、枸杞木虱[17]、梨黑星病、菜青虫、菜蚜等[18]。
目前文献报道的关于苦参碱和氧化苦参碱的痕量检测,主要有液相色谱法[19-20]、电化学法[21],液相色谱-串联质谱法[22-24]、气相色谱-质谱法[25],其中以液相色谱-串联质谱法检测灵敏度高、操作简便,应用最多。基质主要有苦参、蜂蜜、柞蚕等,但鲜有对枸杞基质的考察。同时目前关于苦参碱和氧化苦参碱也基本采用外标法定量,结果准确性受基质效应以及前处理过程中人工操作的影响,定量需要基质曲线校准,方法复杂,准确性较差。本文建立枸杞中苦参碱和氧化苦参碱残留量的液相色谱-串联质谱检测方法,采用同位素内标法定量,简化操作方法,提高结果准确性,以期为开展枸杞中苦参碱和氧化苦参碱检测提供参考。
1 材料与方法
1.1 材料与试剂
枸杞:市售;乙腈、甲醇、甲酸(均为色谱纯):上海安谱实验科技股份有限公司;磷酸(优级纯):国药集团化学试剂有限公司;苦参碱、氧化苦参碱:北京坛墨质检科技有限公司;同位素内标D3-苦参碱、D3-氧化苦参碱:天津阿尔塔科技有限公司。水系微孔滤膜(0.22 μm):海宁市中力过滤设备厂;强阳离子固相萃取柱(SCX,150 mg,6 mL):天津博纳艾杰尔科技有限公司。
1.2 仪器与设备
超高效液相色谱-串联质谱仪(ultra):赛默飞世尔科技(中国)有限公司;高速分散机(T25):德国艾卡公司;涡旋振荡器(MIX-Multi):拓赫机电科技(上海)有限公司;超声清洗器(SB-5200DTD):宁波新芝生物科技股份有限公司;离心机(KL05RF):湖南凯达科学仪器有限公司;纯水机(Milli-Q):美国密理博公司;电子天平(AL204):梅特勒托利多科技(中国)有限公司。
1.3 试验方法
1.3.1 溶液配制
以甲醇配制质量浓度为100 μg/mL 的苦参碱、氧化苦参碱混标溶液,10 μg/mL 的D3-苦参碱、D3-氧化苦参碱混和内标溶液,于-18 ℃下储存。以20% 甲醇水溶液配制1 μg/mL 的混标工作液及混合内标工作液,配制混标溶液1、2、5、10、20 ng/mL 和50 ng/mL 的系列混合标准溶液(内标浓度为25 ng/mL),现配现用。
0.1% 甲酸水溶液:取适量超纯水,加入1.0 mL 甲酸,用水定容至1 L,超声脱气,待用。
1.3.2 样品前处理
1.3.2.1 样品提取
采用高速分散机将枸杞打碎,称取2 g 粉碎试样(精确至0.001 g)于50 mL 塑料离心管中,加入1 μg/mL的混合内标工作液50 μL,加20 mL 1.0% 的磷酸水溶液,涡旋1 min,超声提取30 min,5 000 r/min 离心5 min,收集上清液。
1.3.2.2 净化
强阳离子固相萃取柱依次用6 mL 甲醇和6 mL 水活化,提取液经漏斗过滤后固相萃取柱净化,依次用6 mL 水和6 mL 甲醇淋洗,用10 mL 5% 氨水甲醇溶液洗脱。收集洗脱液,45 ℃氮气吹至近干。加入20% 甲醇水溶液2 mL,涡旋1 min 溶解残余物,水系微孔滤膜过滤,超高效液相色谱-串联质谱仪测定。
1.3.2.3 色谱条件
色谱柱:T3(柱长100 mm,柱内径2.1 mm,填料粒径1.7 μm);流动相A:0.1% 甲酸水溶液,流动相B:甲醇;柱温:30 ℃;进样量10 μL,流速0.3 mL/min。流动相洗脱条件见表1。

表1 流动相及梯度洗脱条件Table 1 Mobile phase and gradient elution conditions
1.3.3 质谱条件
电喷雾离子源(electron spray ionization,ESI);多反应监测(multiple reaction monitoring,MRM);正离子扫描模式;喷雾电压为3 500 V;离子源温度为350 °C。
1.3.4 基质效应
基质效应(matrix effect,ME)按以下公式计算。
式中:M为基质效应;A为基质标准曲线斜率;B为溶剂标准曲线斜率。当ME>0,表明基质效应增强;ME<0,表明基质效应抑制;ME=0 表示不存在基质效应;|ME|<0.2 为弱基质效应;0.2 ≤|ME| ≤0.5 为中等基质效应;|ME|>0.5 为强基质效应。
1.4 数据统计分析
色谱数据分析采用Xcalibur 软件进行处理,回收率试验数据由Office Excel 软件处理,统计图采用Origin Pro 8.5 制作。
2 结果与分析
2.1 前处理条件优化
2.1.1 提取溶剂选择
苦参碱、氧化苦参碱结构见图1。

图1 苦参碱和氧化苦参碱结构Fig.1 Structures of matrine and oxymatrine
由图1 可知,苦参碱由两个哌啶环骈合而成,呈叔胺状态的氮原子处于骈合环之间,立体效应影响较小,氧化苦参碱结构式中含有氮氧化物基团,具有半极性配位键,氧化苦参碱极性大于苦参碱。根据相似相溶原理,应在较大极性的溶剂系统中进行提取。不同提取溶剂对苦参碱与氧化苦参碱峰面积的影响见图2。

图2 不同提取溶剂下苦参碱与氧化苦参碱峰面积比较Fig.2 Comparison of peak areas of matrine and oxymatrine under different extraction solvents
由图2 可知,使用磷酸水溶液作为提取溶液效率较高,这与苦参碱和氧化苦参碱属于强极性物质,应使用极性溶剂提取的结论一致。苦参碱、氧化苦参碱在酸性条件下比较稳定,所以选择磷酸水溶液作为提取溶剂。在不同浓度磷酸水溶液作为提取溶剂时,0.2%、0.5%、1.0% 磷酸水溶液提取后的目标化合物的峰面积随提取溶剂浓度的增加不断增大,1.0% 与2.0% 差别不大。所以选择1.0% 磷酸水溶液作为提取溶剂。
2.1.2 超声时间的选择
不同超声时间,苦参碱与氧化苦参碱峰面积变化见图3。
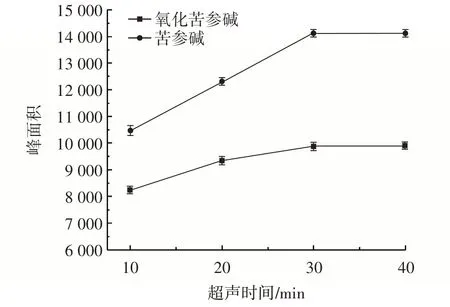
图3 不同超声时间下苦参碱与氧化苦参碱峰面积比较Fig.3 Comparison of peak areas of matrine and oxymatrine under different ultrasound time
由图3 可知,随着超声时间(10~30 min)的延长,目标化合物的峰面积逐渐增大,而在30 min 和40 min 下,目标化合物的峰面积基本没有变化,故选择30 min 作为最终超声时间。超声波可以加速枸杞在溶液中的溶解,提高反应速率和溶解度。但在30 min时,枸杞中的目标化合物已被完全溶解于1.0% 磷酸水溶液中。
2.1.3 固相萃取净化条件选择
试验选择强阳离子固相萃取柱作为净化柱,这是由于苦参碱、氧化苦参碱属于碱性农药,在溶液中带正电荷,强阳离子类的固相萃取柱里有磺酸基团带负电荷,二者形成相互作用力,可以吸附苦参碱和氧化苦参碱。
选择氨水甲醇溶液作为洗脱溶液,是因为二者可提供碱性环境,铵根离子自带正电荷,可与强阳离子萃取柱的负电荷相结合,减弱目标化合物苦参碱、氧化苦参碱与强阳离子萃取柱之间的作用力,使得固相萃取柱吸附的苦参碱和氧化苦参碱解离下来。
试验以目标化合物峰面积为参考,对比不同比例洗脱溶液(甲醇溶液、2% 氨水甲醇溶液、5% 氨水甲醇溶液、10% 氨水甲醇溶液)下对苦参碱、氧化苦参碱的洗脱效果,结果见图4。
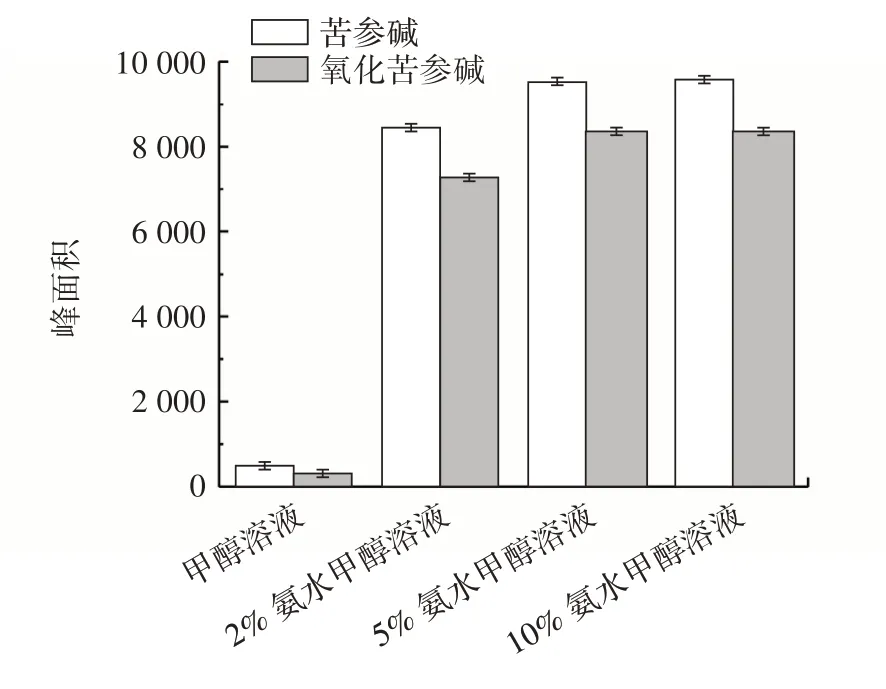
图4 不同洗脱溶剂下苦参碱与氧化苦参碱峰面积比较Fig.4 Comparison of peak areas of matrine and oxymatrine under different elution solvents
由图4 可知,氨水浓度达到5% 以后目标化合物峰面积增加不明显,因此使用5% 氨水甲醇溶液作为洗脱溶剂。继续考察固相萃取柱洗脱曲线,分段收集20 mL 5% 氨水甲醇洗脱液(每2 mL 一段),将洗脱液用氮气吹至近干,1 mL 20% 甲醇溶液复溶,测定每部分苦参碱、氧化苦参碱峰面积,结果见图5。

图5 不同阶段洗脱液下苦参碱与氧化苦参碱峰面积比较Fig.5 Comparison of peak areas of matrine and oxymatrine under different stages of eluent
由图5 可知,10 mL 洗脱液可将目标化合物洗脱完全。
2.2 色谱条件的优化
2.2.1 流动相的选择
试验通过对两种水相溶剂(0.1% 甲酸水溶液、5 mmol 乙酸铵+0.1% 甲酸水溶液)与两种有机相溶剂(乙腈、甲醇)进行交叉比对,分别为0.1% 甲酸水溶液+乙腈、0.1% 甲酸水溶液+甲醇、(5 mmol 乙酸铵+0.1% 甲酸水溶液)+乙腈、(5 mmol 乙酸铵+0.1% 甲酸水溶液)+甲醇。
从响应强度、峰形以及出峰时间3 方面考察,发现0.1% 的甲酸水溶液与甲醇作为流动相效果最明显,离子化效率更高,响应更强,峰形窄而尖锐,保留时间合适。
在等度洗脱模式下,苦参碱、氧化苦参碱和基质、溶剂、杂质总是一同流出,在质谱图上无法区分,同时考虑枸杞样品复杂、对离子源损害较大,决定采用梯度洗脱法将目标物与基质、溶剂、杂质分离。梯度洗脱初始流动相设置水相多而有机相少能将亲水性杂质分离,后逐渐加大有机相比例达到亲水杂质与目标物分离的效果,保留性强的杂质则通过延长洗脱时间去除,最后恢复初始流动相平衡。
2.2.2 色谱柱的选择
试验对比了C18、T3 两种型号色谱柱。目标物在C18 柱下,目标物保留较弱,出峰快,样品中共流出物容易干扰其测定。而在T3 柱下,目标物的峰形及质谱相应强度更好,同时保留时间合适。这是由于目标物属强极性物质,T3 柱是在C18 键合相基础上又加上极性基团修饰,比单独C18 柱对极性分子的保留能力更强,更适合极性较强的目标化合物分析,因此选择T3柱作为苦参碱分析用色谱柱。
2.3 质谱条件优化
试验比较了ESI 负离子和ESI 正离子两种扫描模式下目标化合物的信号强度。配制100 ng/mL 浓度下的4 种混标溶液,并以10 μL/min 的流速直接注入质谱。结果表明,目标化合物在正离子扫描模式下的信号强度明显高于负离子扫描模式下,这是由于苦参碱、氧化苦参碱含有氮元素,容易结合得到一个质子形成正离子,因此采用ESI 正离子扫描模式。进一步优化ESI 正离子扫描模式下MRM 的母离子、子离子、碰撞能量和去簇电压,每个目标物选择丰度较高、干扰最少的2 组离子对用于MRM 监测。最终优化的质谱条件如表2 所示。优化后苦参碱、氧化苦参碱及各自同位素内标MRM 检测见图6。

图6 4 种化合物多反应监测(MRM)色谱Fig.6 Chromatograms of four compounds in MRM mode

表2 MRM 模式下的质谱参数Table 2 Mass spectrometry parameters in MRM mode
2.4 基质效应评价
在液质检测中,基质效应普遍存在,影响定量结果的准确性,而同位素内标的引进在一定程度上可以消除基质效应,提高定量准确度。在使用外标法时结果显示|ME|苦参碱=0.17,|ME|氧化苦参碱=0.18,苦参碱与氧化苦参碱呈现弱基质效应。内标法引入后|ME|苦参碱=0.04,|ME|氧化苦参碱=0.03,内标的引入很大程度减小了基质效应,可以采用溶剂配制工作曲线,简化了定量操作过程。
2.5 方法学验证
2.5.1 线性方程与检出限、定量限
以3 倍信噪比确定方法检出限,以10 倍信噪比确定定量限,结果如表3 所示。
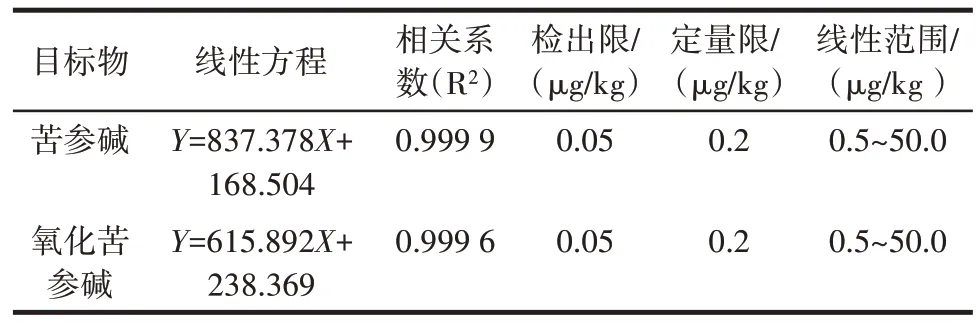
表3 苦参碱和氧化苦参碱的线性关系及检测灵敏度Table 3 Linear relationship and detection sensitivity of matrine and oxymatrine
2.5.2 回收率与相对标准偏差
取空白样品,选择低、中、高3 个浓度进行添加回收试验,重复试验6 次,回收率及相对标准偏差如表4所示。

表4 方法回收率和相对标准偏差Table 4 Method recovery rate and RSD
2.6 实际样品测试
市场上采集15 份枸杞样品,经测定,有2 份样品检出氧化苦参碱成分,分别为0.76、0.83 μg/kg,其余均为未检出。GB 2763—2021《食品安全国家标准食品中农药最大残留限量》仅制定了部分蔬菜水果的苦参碱限量标准(1~5 mg/kg),检出苦参碱成分的枸杞样品远未达到此限量,枸杞中苦参碱对人可能产生的风险较小。同时由于样品量较小,可能代表性不够,下一步计划扩大样品采样继续检测。
3 结论
本文采用液相色谱-串联质谱检测枸杞中苦参碱和氧化苦参碱,通过优化提取溶液、超声时间、洗脱溶液、洗脱溶液体积等前处理条件达到痕量检测的目的,同时通过查阅相关标准及文献,首次采用同位素内标法定量,该方法简便快速、结果准确性高,大大降低了基质效应以及人工操作误差,适合于枸杞中苦参碱和氧化苦参碱的测定。
猜你喜欢
中国油脂(2020年3期)2020-04-10 02:08:54
中成药(2018年12期)2018-12-29 12:25:54
天然产物研究与开发(2018年5期)2018-06-13 03:23:34
无机化学学报(2016年8期)2016-12-06 09:05:14
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:57
中学生理科应试(2016年2期)2016-05-30 14:23:28
广东石油化工学院学报(2016年3期)2016-05-17 05:16:21
中国民族医药杂志(2016年5期)2016-05-09 07:43:50
化学分析计量(2016年1期)2016-03-14 00:35:19
分析测试学报(2015年3期)2016-01-13 06:18:20
