
超高效液相色谱-串联质谱测定水源水中176 种药品和个人护理用品含量
2024-03-10 11:25:24吴少明刘文菁何孟杭陈言凯詹重清欧阳立群
食品科学 2024年4期
吴少明,刘文菁,戴 明,何孟杭,陈言凯,王 征,詹重清,欧阳立群
(福建省产品质量检验研究院,国家加工食品质量检验检测中心,福建 福州 350002)
药品和个人护理用品(pharmaceutical and personal care products,PPCPs)是一类具有伪持久性的“新兴环境污染物”[1],包括各种抗生素、性激素、兴奋剂、退烧药、降压药、减肥药、避孕药、麻醉剂等,涉及生活及生产的各个方面[2],近年来在国内外备受关注。PPCPs主要通过生活中的废水进入环境中,会对生态环境和人类健康造成潜在危害[3-6]。为此,澳大利亚、欧盟、美国国家水研究所相继出台了相关标准,对包括双氯芬酸在内的PPCPs进行了水质指标的浓度限定[7];美国食品药品监督管理局于2016年明确禁止在抗菌清洁类产品中添加三氯生[8],我国新颁布的GB/T 5750—2023《生活饮用水标准检验方法》也新增了39 项PPCPs的检测指标。
随着仪器分析技术的不断发展,全球范围内已经发现PPCPs存在于各地区水体中,包括美国[9]、意大利[10]、非洲[11]、伊朗[12]和中国[13]等地,水平从ng/L到μg/L不等。Kolpin等[14]在美国139 条河流中发现了85 种PPCPs药物;McArdell等[15]对3 座污水处理厂和Glatt河流中的大环内酯类物质进行调查,发现了不同浓度的克拉霉素;Miao Xiusheng等[16]调查了加拿大5 座城市8 个污水处理厂出水中的31 种抗生素残留情况,发现除磺胺甲恶唑,还检测到磺胺吡啶等新药物残留。Nakada等[17]对日本利根川的药物残留情况进行调查,发现在70 种检测的药物中有57 种被检出,包括克拉霉素、咖啡因、苯扎贝特和卡马西平等。Murata等[18]则研究了12 种抗生素在37 条河流中的污染状况,发现这些河流中的抗生素总质量浓度范围为未检出至626 ng/L,中位数为7.3 ng/L;Yoon等[19]对韩国汉江中31 种内分泌干扰物和PPCPs的分布情况进行调查,发现碘普罗胺、萘普生、避蚊胺、卡马西平、咖啡因等化合物在河流和小溪样品中经常被检测到;Sun Qian等[20]研究福建九龙河PPCPs化合物的空间分布及趋势,发现冬季的河水样品中解热镇痛药和消炎药的检出率均高达100%,其中扑热息痛、布洛芬、酮洛芬、双氯芬酸的检出浓度较高;甚至在我国一些地区的居民饮用水中甚至也检测到PPCPs的存在,芜湖、珠三角和苏州等地的居民饮用水中均检出多类抗生素[21-23],说明PPCPs在水中的存在已是不争的事实。因此,建立一种准确性好的高通量检测方法,对了解我国水体中PPCPs的污染水平、分布特征和迁移转化规律具有重要意义。
目前,液相色谱-串联质谱技术是PPCPs测定最常用的方法[2,13,21-23],但现阶段的检测技术主要是针对同类或少数种类PPCPs,对多类PPCPs的同时检测技术报道较少,且通常需要多种前处理方法才能完成多项目的测定。因此,本研究旨在利用超高效液相色谱-串联质谱(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)的高准确性和高通量优势,建立仅需一次前处理和一次上机测试即可完成水源中20大类共176 种PPCPs的快速筛查和准确定量分析方法,涉及的化合物主要包括文献报道中常检出以及日常生活中可能用到的PPCPs,并将其应用于对福州水源水中PPCPs的筛查和定量分析,该技术可为水源水中的安全监测、预警和执法提供一定的技术支持。
1 材料与方法
1.1 材料与试剂
20大类共176 种PPCPs液体混合标准溶液(每一类为1 瓶混合标准溶液,质量浓度均为100 μg/mL)天津阿尔塔公司;甲酸、乙腈、甲醇(均为色谱纯)德国Merck 公司;冰乙酸、二甲基亚砜(dimethyl sulfoxide,DMSO)、氨水(均为分析纯)上海国药集团化学试剂有限公司;Cleanert PEP固相萃取柱(6 mL/500 mg)美国Agela公司;UPLC HSS T3色谱柱(2.1 mm×150 mm,1.8 μm)美国Waters公司;实验用水为屈臣氏蒸馏水。
1.2 仪器与设备
1290超高效液相色谱仪 美国Agilent公司;Triple Quad 5500三重四极杆串联质谱仪 美国AB SCIEX公司;Multifuge×4R Pro高速冷冻离心机 美国Thermo Fisher公司;DS-8510 DTH超声波振荡器 上海分析仪器有限公司;MS 3 basic旋涡振荡器 德国IKA公司;AutoEVA-20Plus全自动平行浓缩仪 天津睿科仪器有限公司。
1.3 方法
1.3.1 水样采集
选用福州市闽江水源水,从西头闽侯至东头马尾区,每隔5 km设置一个采样点,共计13 个采样点,每个采集点采集2 个样品,共26 个水样。水样的采集和保存按照GB/T 5750.1—2006《生活饮用水标准检验方法》。将采集的水样密闭保存于棕色玻璃瓶中,采样量为500 mL,采集后24 h内进行实验。
1.3.2 样品前处理
准确取水样50 mL于离心管中,用5%甲酸或5%氨水溶液调节pH值为7,转移至事先用6 mL甲醇和6 mL水活化的Cleanert PEP固相萃取柱中(加装50 mL大体积管),待其自然重力作用流出后,用6 mL蒸馏水淋洗小柱,弃去全部淋洗流出液,减压抽干后先以5 mL 50%(体积分数,后同)甲醇-乙腈溶液洗脱,再以5 mL含1%甲酸的50%甲醇-乙腈溶液洗脱,收集两次洗脱液,并加入100 μL DMSO后于平行浓缩仪中40 ℃水浴浓缩至近干,以40%甲醇溶液定容至1.0 mL,涡旋混匀后过0.22 μm聚四氟乙烯(polytetrafluoroethylene,PTFE)滤膜,上机测试。
1.3.3 标准溶液及标准工作曲线的制作
准确吸取20 大类标准溶液(质量浓度均为100 μg/mL)各1.00 mL至100 mL容量瓶中,以甲醇稀释至刻度,配制成质量浓度均为1.00 μg/mL的混合标准中间液,转入棕色瓶中,于-18 ℃保存。分别准确吸取适量混合标准中间液,以40%甲醇溶液稀释成质量浓度分别为5.00、10.0、20.0、50.0、100、200 ng/mL,绘制标准曲线。
1.3.4 仪器条件
1.3.4.1 色谱条件
色谱柱:UPLC HSS T3色谱柱(2.1×150 mm,1.8 μm);流动相:水相(A)为0.1%(体积分数)甲酸水(含0.2 mmol/L乙酸铵)溶液,有机相(B)为甲醇;流速为0.3 mL/min,柱温为40 ℃,进样体积为5 μL,洗脱程序为:0~6 min,95%~5% A、5%~95% B;6~9 min,5% A、95% B;9~9.01 min,5%~95% A、95%~5% B;9.01~12 min,95% A、5% B。
1.3.4.2 质谱条件
电喷雾电离源(electro-spray ionization,ESI):正离子、负离子切换扫描模式;多反应监测扫描,分段扫描方式(分段时间段为各化合物保留时间左右各30 s);离子化电压:正离子模式5500 V,负离子模式4500 V;雾化气压力55 psi;辅助气压力55 psi;气帘气压力30 psi;离子源温度550 ℃;定性离子对、定量离子对及其他质谱参数见表1。

表1 176 种PPCPs化合物的质谱参数Table 1 Mass spectrometric parameters for the detection of 176 PPCPs
1.4 数据处理与分析
采用Multiquant 4.0软件进行数据处理及分析,采用Excel 2016软件作图。
2 结果与分析
2.1 仪器条件的优化
2.1.1 质谱条件的优化
用50%甲醇溶液将标准溶液稀释至质量浓度均为200 ng/mL的混合标准使用液,采用外置针泵连续进样模式、ESI源进行采集分析。根据化合物的性质,分别采用对应的离子扫描模式(正、负),先采用Q1(Q1Scan)扫描,分别获得[M+H]+或[M-H]-的母离子,再进行MS2子离子扫描,得到每个化合物的主要碎片离子,选取相对丰度最高及次高的碎片离子作为定量离子和定性离子,以满足欧盟2002/657/EC的规定,即保证检测需达到4 个确证点的要求。再分别进行多反应监测扫描,优化各离子对的去簇电压、碰撞能,最终得到161 种化合物采用ESI+模式,其余15 种化合物则采用ESI-模式,优化后的质谱参数见表1。
2.1.2 色谱条件的优化
对于PPCPs的测定,文献中大多采用的是C18色谱柱。本实验对比了常用的Acuity UPLC BEH C18(150 mm×2.1 mm,1.7 µm)和Acuity UPLC HSS T3(150 mm×2.1 mm,1.8 µm)色谱柱分析效果。结果发现,在相同流动相条件下,间氨基苯甲酸、普鲁卡因胺、沙丁胺醇、阿莫西林等强极性的化合物,在HSS T3色谱柱上得到更好的保留,三氯卡巴、炔诺肟酯、丙酸睾丸素等极性较弱的目标物也不会过强地保留,整体表现出更尖锐的峰形,且可以耐受更高比例的水相,因此选择Waters Acuity UPLC HSS T3作为分析柱。此外,对比了0.1%甲酸-甲醇、0.1%甲酸-乙腈、0.1%甲酸(含0.02 mmol/L乙酸铵)-甲醇、0.1%甲酸(含0.02 mmol/L乙酸铵)-乙腈4 种流动相的效果。结果显示,有机相为甲醇时,可以提高可的松、咖啡因、青霉素G等大多数化合物的响应,但炔诺肟酯、地尔卓卡、替米考星等化合物色谱峰出现分叉;而含有乙酸铵时,色谱峰分叉的情况得到明显改善,这是由于大多数化合物采用的是ESI+模式,甲醇是质子供体,能够提高目标物的质子化,而乙酸铵是流动相改进剂,可以有效改善峰形。因此,选择0.1%甲酸(含0.02 mmol/L乙酸铵)-甲醇作为流动相,176 种化合物的提取离子色谱图见图1。

图1 176 种化合物的提取离子色谱图(20 ng/mL)Fig.1 Extracted ion chromatograms of 176 PPCPs (20 ng/mL)
2.2 净化条件的优化
2.2.1 水样pH值的选择
对于水中PPCPs的测定,研究中通常采用HLB[24-26]、PEP-2[27-28]固相萃取柱净化富集,并认为不同化合物的回收率和样品pH值有关[27-28]。本实验选取Cleanert PEP固相萃取柱作为净化柱,对比了不同pH值(3、5、7、9、11)的水样进行上样以及淋洗步骤,收集流出液进行后续实验,研究PPCPs化合物的损失情况,平行实验n=2。结果发现,水样pH值低于9时,所有化合物均未损失;而pH值为9时,12 种化合物有不同程度的损失,尤其是对氨基苯甲酸(损失率为93.6%)、间氨基苯甲酸(损失率为97.4%)、磺胺醋酰(损失率为72.1%);而当水样pH值达到11时,17 种化合物均有不同程度的损失,其中10 种化合物损失率高于87.4%(图2),说明水样为碱性条件时,一些化合物在上样或淋洗过程存在损失,综合考虑选择水样pH值为7。

图2 样品pH值对上样和淋洗过程中PPCPs损失率的影响Fig.2 Effect of sample pH on the loss rates of PPCPs during loading and washing
2.2.2 洗脱溶剂的选择
Cleanert PEP固相萃取柱填料是由聚苯乙烯和二乙烯苯组成的,同时具有亲水性和憎水性基团,对各类强极性、弱极性的化合物均具有较好的吸附,而常用的洗脱溶剂为甲醇和乙腈。但本研究涉及176 种PPCPs,种类繁多且性质差异较大,考虑到洗脱溶剂酸碱性可能对结果产生影响,分别对比甲醇、乙腈、50%甲醇-乙腈、含1%甲酸的50%甲醇-乙腈及含1%氨水的50%甲醇-乙腈作为洗脱溶剂的效果。结果见图3,采用甲醇洗脱时,54.0%(95/176)的化合物回收率高于60%;乙腈洗脱时,67.0%(118/176)的化合物回收率高于60%;50%甲醇-乙腈洗脱时,72.2%(127/176)的化合物回收率高于60%;1%甲酸化的50%甲醇-乙腈或1%氨水的50%甲醇-乙腈洗脱时,85.8%(151/176)的化合物回收率高于60%,说明酸性或碱性的50%甲醇-乙腈洗脱效果较好;此外,采用1%甲酸化的50%甲醇-乙腈效果较差的25 种化合物中,50%甲醇-乙腈洗脱效果均较好,回收率在68.8%~129.6%之间,继续实验,先采用5 mL 50%甲醇-乙腈洗脱,再加入5 mL 1%甲酸化的50%甲醇-乙腈继续洗脱,结果发现176 种化合物的回收率均较为满意,回收率在73.2%~123.2%之间。因此,选择先以5 mL 50%甲醇-乙腈洗脱,再以5 mL 1%甲酸化的50%甲醇-乙腈洗脱的分步洗脱方式。

图3 不同洗脱溶剂洗脱后回收率高于60%的化合物占比Fig.3 Compounds percentage with recoveries higher than 60% with different elution solvents
2.3 浓缩条件的选择
2.3.1 浓缩温度的选择
由于净化后的溶液体积较大,且洗脱液为高比例有机相,与流动相初始比例差距较大,会产生溶剂效应,需将净化液浓缩后进行溶剂转换。但由于176 种PPCPs化合物种类繁多,性质差异较大,可能存在热不稳定的化合物。因此,以50%甲醇-乙腈为溶剂配制质量浓度均为50 ng/mL的标准溶液,采用平行浓缩仪进行浓缩,分别对比浓缩温度为30、40、50、60 ℃条件下对目标物回收率的影响。如图4所示,当浓缩温度到达40 ℃时,头孢拉定的回收率下降12.1%;当温度达到50 ℃时,9 种化合物的回收率明显下降,降至32.5%~78.8%;而当温度达到60 ℃时,25 种化合物的回收率下降明显,仅为1.7%~67.3%(图4);除图中的化合物外,其余化合物并没有明显损失,回收率均在90%以上,但考虑到30 ℃时浓缩时间较长(约60 min),最终选择浓缩温度为40 ℃。

图4 浓缩温度对PPCPs回收率的影响Fig.4 Effect of condensation temperature on the recovery rates of PPCPs
2.3.2 DMSO用量的选择
为了考察经浓缩吹干后PPCPs的回收效果,以50%甲醇-乙腈配制质量浓度为100 ng/mL的标准溶液,置于平行浓缩仪中40 ℃氮气吹干,结果发现氯霉素、甲砜霉素、氟甲砜霉素、甲硫威、地尔硫卓、瑞格列奈6 种化合物无明显损失,回收率在93.2%~102.1%之间,而其余回收率均较低,回收率均低于80%,其中103 种化合物回收率低于60%,这是由于吹干后,一方面10%乙腈无法完全溶解目标物导致损失;另一方面,氮吹干后一部分目标物随溶剂共同挥发或者分解导致。而在进行大批量样品处理时很难避免每个样品都不被吹干,吴少明等[29]认为DMSO与水、甲醇、乙腈等都互溶,且对于PPCPs化合物均有较好的溶解性,且沸点较高(189 ℃),在浓缩前添加一定量DMSO可能会减少目标物的损失,但过量的DMSO可能会对仪器造成损坏。因此,本实验通过选择10、20、50、100、200 μL DMSO进行测定,结果发现,随着DMSO添加量的增多,所有化合物的回收率逐渐升高,在100 μL时基本无损失,每个化合物回收率均为86.6%~104.7%之间,符合分析要求,因此,选择DMSO 用量为100 μL,并对比该用量下添加DMSO与未添加组对经浓缩吹干后的176 种PPCPs的回收率影响,结果见图5。
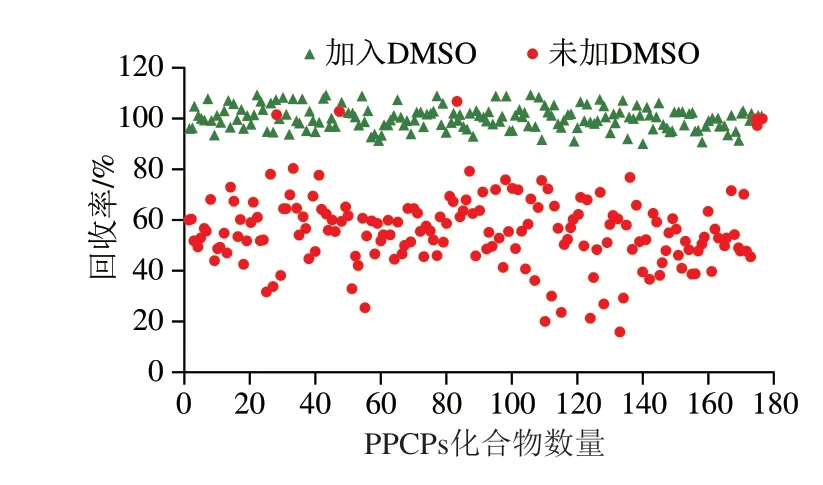
图5 添加100 μL DMSO对PPCPs回收率的影响Fig.5 Effect of the addition of 100 μL of DMSO on the recovery rates of PPCPs
2.4 滤膜的选择
常用的滤膜主要有尼龙滤膜和PTFE滤膜,相较PTFE滤膜,尼龙膜吸附能力更强,得到的滤液更干净,但也可能吸附目标物。本实验以40%甲醇溶液配制标准品溶液质量浓度为20 ng/mL,分别使用0.22 μm尼龙滤膜和0.22 μm PTFE滤膜过滤,对比两种滤膜处理后对回收率的影响。结果发现,采用PTFE膜对176 种化合物均没有明显的吸附,回收率均在97.0%以上;而尼龙膜对29 种化合物存在不同程度的吸附,13 种化合物回收率降至3.0%~67.6%之间,而其余16 种化合物则没有回收(图6),说明尼龙膜吸附较严重,故选择0.22 μm PTFE滤膜过滤。

图6 尼龙滤膜对PPCPs的吸附Fig.6 Adsorption efficiencies of PPCPs on nylon filter membrane
2.5 方法学评价
2.5.1 线性范围和定量限
将1.3.3节配制的标准系列工作曲线上机测定,以各目标物峰面积为纵坐标(Y),各自质量浓度为横坐标(X)绘制标准曲线,176 种目标物在5.00~200 ng/mL质量浓度范围内线性关系较好,相关系数r>0.99。选取阴性水样进行加标回收实验,由于样品为水,信噪比较大,若以3 倍信噪比和10 倍信噪比确定检出限和定量限,所得结果大部分较小。因此,本实验以各目标物在线性范围内满足实际回收率在60%~130%之间的最低加标水平为定量限,最终确定176 种PPCPs化合物的定量限均为0.1 ng/mL。
2.5.2 准确度、精密度
以阴性水样进行0.1、0.4、1.0 ng/mL 3 种水平的加标回收实验,每个加标水平平行实验6 次,计算各自的加标回收率和相对标准偏差(relative standard deviation,RSD)。结果显示,176 种化合物的平均回收率在68.0%~126.7%之间,RSD在1.1%~10.3%之间。此外,在同样条件下,将同一加标水平(0.5 ng/mL)的样液每隔2 h进样一次,连续进样6 次,以6 次的回收率RSD计算日内精密度;并连续6 d进行同水平(0.5 ng/mL)加标实验,以6 d 所得回收率的RSD计算日间精密度。结果表明,176 种PPCPs化合物的日内精密度为2.2%~5.7%,日间精密度为3.2%~9.8%。说明该方法具有较好的准确度与精密度,满足GB/T 27404—2008《实验室质量控制规范食品理化检测 附录F》的要求。
2.6 实际样品测定
采用新开发的方法测定13 个闽江水源水样,共检出11 种PPCPs化合物,其中咖啡因质量浓度为13.5~58.9 ng/mL、1,7-二甲基黄嘌呤质量浓度为1.5~7.5 ng/mL、扑热息痛质量浓度为0.6~6.3 ng/mL、缬沙坦质量浓度为1.3~4.3 ng/mL、头孢拉定质量浓度为0.5~2.5 ng/mL、磺胺质量浓度为ND~1.0 ng/mL、甘宝素质量浓度为ND~1.0 ng/mL、替米沙坦质量浓度为ND~0.9 ng/mL、西咪替丁质量浓度为ND~0.8 ng/mL、雷尼替丁质量浓度为ND~1.2 ng/mL、三氯卡巴质量浓度为ND~0.2 ng/mL,其余化合物均未检出。
3 结论
本研究对仪器条件以及前处理条件进行系统优化,建立了一种Cleanert PEP固相萃取小柱净化-超高效液相色谱-串联质谱法同时测定水中176 种PPCPs化合物含量的高通量分析方法。方法学验证表明该方法具有良好的线性范围、较低的检出限、较高的准确度和精密度。该方法只需一次前处理以及一次上机测试即可完成176 种PPCPs化合物的分析,涉及的化合物更多,种类更齐全,可为水环境中PPCPs化合物含量的监管提供技术支持。
猜你喜欢
陶瓷学报(2020年3期)2020-10-27 02:08:12
辐射防护通讯(2019年3期)2019-04-26 05:16:26
Clinical Research Communications(2019年1期)2019-04-23 07:30:46
绿色科技(2018年24期)2019-01-19 06:36:50
意林(儿童绘本)(2018年10期)2018-11-08 11:01:36
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
应用化工(2014年1期)2014-08-16 13:34:08
无机化学学报(2014年3期)2014-02-28 17:30:58
河南科技(2014年12期)2014-02-27 14:10:32
