
微波消解-电感耦合等离子体发射光谱(ICP-OES)法测定固体中兽药制剂中金属元素
2024-03-04 00:49刘占通张崇威
中国无机分析化学 2024年4期
刘占通 张崇威 韩 立 李 娜
(河南省农畜水产品检验技术研究院,郑州 450008)
2019年农业农村部公告第194号规定,除中药和抗球虫外,养殖环节禁止使用所有促生长类药物,自此药物饲料添加剂退出历史舞台。而中兽药因其低毒、无药残、无耐药性、价格低廉、符合法规要求等优势[1],在兽药市场脱颖而出。中兽药的崛起,势必给行业带来商机,但检测手段的滞后,使得不法分子有机可乘。通过添加违禁物质,特别是具有特定作用的金属元素,以达到非法获利的目的。例如,养殖环节增加铜、锌的摄入可促进动物生长、改善动物皮毛颜色[2],增加有机铬和有机砷的摄入,可刺激动物生长、改善肉质并具有杀菌作用[3-4],锰元素是动物生长不可或缺的微量元素,特别是母畜缺乏会导致很多疾病,硒元素的刻意摄入,提高了动物食品中硒元素含量,更是迎合了越来越受到热捧的富硒食品。而钾、钠、镁、钙、磷等常量元素在动物的生长过程中更是不可或缺。但金属元素的长期摄入,将通过食物链富集,最终对土壤环境及人体健康造成影响[5-6]。其中 Fe、Ca、Se、K、P等属于有益元素[7],但长期过量使用慢性中毒,铁过量会造成运动失调、肌肉痉挛及呼吸困难,硒过量会造成贫血、关节强直,锌过量会造成贫血、生长发育迟缓等[8]。Pb、Cr、Cd、As、Cu等重金属元素的过量摄入会对肝脏造成毒害,甚至引发各类病变乃至癌症[9-10]。但国家对中兽药中金属元素的含量未做明确规定。因此,建立中兽药中金属元素检测方法显得十分必要。
目前,金属元素检测前处理消解方法主要有湿法消解、干法消解和微波消解[11-12]。微波消解具有操作简单、成本低、空白值低等优点,逐渐成为主流消解方式[13-14]。仪器方法主要有原子吸收光谱法[15-17]、原子荧光光谱法[18-19]、电感耦合等离子体发射光谱法(ICP-OES)[20-23]、电感耦合等离子体质谱法(ICP-MS)[24-27]。原子吸收光谱仪及原子荧光光谱仪灵敏度高,但不能同时进行多元素检测。ICP-OES及ICP-MS均具有灵敏度高、检出限低,分析速度快,多元素同时检测等优点[28],但ICP-OES在线性宽度及受基体干扰方面优于ICP-MS,且能同时测定微量元素及常量元素而不产生污染。目前,对于中兽药中金属元素检测的ICP-OES方法还未有报道。因此,通过对样品前处理及仪器参数的比较研究,建立一种高效、便捷、准确测定中兽药中多金属元素的分析方法具有现实意义。
1 实验部分
1.1 材料与试剂
Cr、As、Cd、Pb、Na、Mg、K、Mn、Fe、Cu、Zn、Se、Ca、P单元素储备标准溶液(1000 μg/mL)购自中国计量科学研究院,Ni内标溶液(100 μg/mL)购自国家有色金属及电子材料分析测试中心。
60%硝酸(优级纯,德国Merck公司);过氧化氢(优级纯,天津市科密欧化学试剂有限公司);水为超纯水(18.2 MΩ·cm),由Milli-Q超纯水系统制得。
1.2 仪器与设备
PQ9000电感耦合等离子体光谱仪(德国耶拿分析仪器有限公司)、AB204-N电子分析天平(梅特勒-托利多(上海)有限公司)、EHD-40赶酸仪(北京东航科仪仪器有限公司)、MARS 6 Classic微波消解仪(美国CEM公司)。
1.3 实验方法
1.3.1 样品来源
黄芩颗粒、益母生化散、麻杏石甘散、扶正解毒散、七清败毒颗粒5种固体中兽药制剂均来自市场随机采购,包装完好。为防止污染,样品不做处理,实验时直接称取样品。
1.3.2 样品前处理
取固体中兽药制剂约0.5 g(精确至0.001 g)于消解罐中,加6 mL硝酸及1 mL过氧化氢,盖内盖后静置过夜。置微波中消解后冷却至室温,置赶酸仪上赶酸,赶酸仪温度从室温升至170 ℃,并在170 ℃保持1.5 h,将消解液赶至约0.5 mL,冷却至室温,用水分3~5次清洗内壁并转移清洗液至25 mL量瓶中,加适当浓度内标溶液,用水定容至刻度。同时做试剂空白实验。
1.3.3 标准工作溶液配制
以3%硝酸溶液为介质,按照表1所示,对各元素浓度梯度进行逐级稀释,作为标准工作溶液,同时加入Ni标准溶液(100 μg/mL)作为各元素内标溶液,内标溶液上机浓度为100 μg/L。

表1 14种元素在标准系列溶液中的浓度Table 1 Concentration of 14 elements in standard series solutions
1.3.4 仪器条件
微波消解程序升温参数:第一步为经15 min从室温升至150 ℃并保持5 min;第二步经3 min从150 ℃升到170 ℃,保持5 min;第三步经3 min从170 ℃升到190 ℃,保持25 min。ICP-OES主要工作参数见表2。

表2 ICP-OES主要工作参数Table 2 Working parameters of ICP-OES
2 结果与讨论
2.1 消解方式的选择
检测金属元素的样品消解方式一般有4种:湿法消解、干法消解、高压消解和微波消解。通过对前处理消解方式的比较,结果表明:湿法消解设备简单,操作容易,但易造成污染,受实验人员操作影响较大,精密度差;干法消解温度一般为500~600 ℃,造成易挥发元素损失,检测结果偏低,如铅、硒、砷、隔等元素;高压消解法操作简便,重现性好,酸用量少使得空白本底值较低,但在消解有机物含量高或者消解酸体系中含有高氯酸时易发生爆炸;微波消解法是在密闭空间进行消解,有效防止了交叉污染,减少挥发性元素损失,且实现自动化操作。本实验比较了湿法消解酸体系为硝酸-硫酸(1∶1),高压消解酸体系为硝酸-高氯酸,微波消解法酸体系为硝酸-过氧化氢(6∶1)以及干法消解,对固体中兽药制剂做5平行阳性添加并进行消解处理,经ICP-OES测定,扣除本底后对各元素平均回收率进行结果比较,如图1所示。
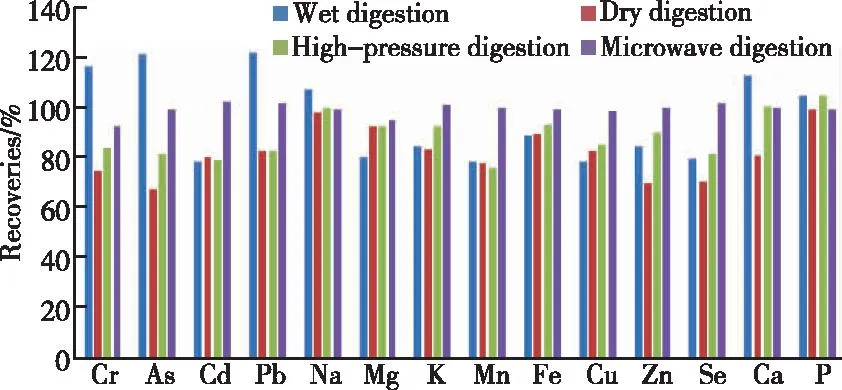
图1 不同前处理方式对各元素平均回收率的影响Figure 1 Effects of different pre-treatment methods on the average recoveries of each element.
由图1可见,微波消解法前处理对大部分元素回收率明显优于其他3种前处理结果,对固体中兽药制剂中多种金属元素同时检测具有明显优势。
2.2 微波消解参数选择
因硝酸具有强酸性及强氧化性,是微波消解的主要用酸,常用消解液体系为硝酸-盐酸、硝酸-氢氟酸、硝酸-高氯酸,同时加入过氧化氢促进氧化。盐酸的氧化性较弱,分解有机物能力差,且对后续的测定造成干扰[29];高氯酸在高温高压下容易与有机物中羟基反应生成不稳定高氯酸酯发生爆炸[30];而氢氟酸一般针对硅酸盐基质的消解[31],且对仪器部件如石英雾化室、雾化器和矩管有腐蚀作用。因此,实验采取仪器厂家推荐两种方法进行比较,即5 mL硝酸(60%)、6 mL 硝酸(60%)、6 mL硝酸(60%)+1 mL过氧化氢。同时分别设170、180、190 ℃ 3个最高消解温度,分别加酸经消解、赶酸、转移并定容后,做正交实验。结果表明:温度为170或180 ℃时,消解后溶液均呈现不同程度的浑浊,如图2中①所示;消解温度为190 ℃时,添加5 mL硝酸微波消解后消解液有浑浊,如图2中②所示;添加6 mL硝酸微波消解后消解液无浑浊,但颜色显黄色,如图2中③所示;添加6 mL硝酸+1 mL过氧化氢后消解液清澈透明,无固体杂质,结果如图2中④所示。

图2 不同消解液体系消解后消解溶液状态Figure 2 Digestion solution status of different digestion liquid systems after digestion.
由图2可见,消解体系为6 mL硝酸+1 mL过氧化氢时,消解效果理想。若添加消解液后静置过夜再进行消解,不但效果更佳,还可有效防止爆管现象。
2.3 赶酸温度的选择
进样溶液酸度过高,易造成进样管等仪器部件的过早老化,且基质效应显著[32],因此,应严格控制进样溶液中酸的浓度。赶酸温度的高低对于挥发性元素特别是As的回收率有显著影响,温度高于190 ℃赶酸2 h对As回收率有明显影响,回收率为82%。考察试样经消解后置入赶酸仪中从室温升至130、150、170、190、210 ℃ 5个温度点,赶酸至消解液约0.5 mL,各赶酸温度对应As回收率如图3所示。

图3 赶酸温度对As回收率的影响Figure 3 Effect of acid driving temperature on As recovery.
由图3可见,赶酸温度为130~170 ℃时,As回收率无明显变化,当赶酸温度升至190 ℃时,As回收率显著下降,因此,为保证挥发性元素的准确测定及提高检测效率,将赶酸温度设置为170 ℃。
2.4 分析谱线波长的选择
PQ9000电感耦合等离子体光谱仪针对测定的每种元素,自带多行业多种推荐特征谱线。选择特征分析谱线应综合考虑谱线灵敏度、共存元素干扰、背景干扰及该元素线性范围[33],遵循检出限低、灵敏度高、干扰小、信噪比高的原则。实验选取每种元素推荐的食品行业2~3条谱线,比较各谱线样品溶液与标准溶液峰形,选取无杂峰干扰、灵敏度高的分析谱线,并选择合适的背景点扣除背景。通过分析,最终确定14种元素的分析线如表3所示。

表3 待测元素分析谱线Table 3 Analysis lines for elements to be tested
2.5 方法可行性验证
实验通过试剂加标回收、进样前样品溶液加标并扣除本底后回收验证前处理方法及仪器条件的可行性。各元素回收率及精密度见表4。

表4 各阳性添加回收率及精密度比较Table 4 Comparison of recoveries and precision for each positive addition(n=5)
由表4可知,各方式加标回收率均在90.0%~110%,无明显差异。试剂添加回收率与样品处理液添加回收一致,说明前处理过程中杂质去除干净,有机物消解完全,待测元素损失少,仪器条件设置恰当。
2.6 线性方程与灵敏度
按照设定的仪器参数条件,对各元素标准系列浓度点进行测定,以各元素仪器响应信号强度与Ni标液仪器响应信号强度比值为纵坐标,各元素浓度点为横坐标,进行线性拟合。同时对10份样品空白按照实验方法进行前处理并测量,以10次空白溶液各元素响应强度与Ni标液响应强度比值所对应的元素含量的3倍标准偏差为方法检出限[34]。线性结果如表5所示。

表5 各元素线性方程及检测限Table 5 Linear equation and detection limit of each element
2.7 实际样品检测
采用上述检测方法对市场上随机购买的黄芩颗粒、益母生化散、麻杏石甘散、扶正解毒散、七清败毒颗粒5种固体中兽药制剂进行元素分析,结果见表6。

表6 样品元素分析结果Table 6 Analysis results of elements in samples /(mg·kg-1)
结果显示,不同种类中兽药的不同金属元素含量差别较大,即使同一金属元素差别也较明显,如重金属As及Mg、Mn、Ca元素最大相差约100倍,Cr及Pb相差约200倍,Fe最大相差大于300倍,Cu、Zn最大相差也在30倍以上,这与中兽药的生长环境、加工炮制工艺紧密相关[35],但不排除为达到经济效益人为因素的添加。随着全国兽用抗菌药使用减量化行动的实施,中兽药是替抗减抗的首选药物,中兽药质量事关畜产品质量安全,因此中兽药中金属元素的含量测定方法研究至关重要。
3 结论
建立了微波消解-ICP-OES同时测定固体中兽药制剂中常见的14种常量元素及痕量元素的方法,通过对消解方式的选择、消解酸体系及消解温度、赶酸温度的优化,分析谱线波长的选择,最大程度地去除杂质,提高了分析元素检测灵敏度,各元素检出限在0.1~1.0 mg/kg,并提出了试剂空白添加、样品空白添加、空白样品处理液添加相比较以判断前处理效果的方法,方法回收率在90.0%~110%,RSD均小于10%,具有较好的准确度和精密度。方法前处理操作简便,成本低、效率高,为固体中兽药制剂中同时测定多种元素提供参考。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
云南化工(2021年7期)2021-12-21
云南化工(2021年8期)2021-12-21
中学生数理化·高一版(2020年11期)2020-12-14
中成药(2018年11期)2018-11-24
电镀与环保(2017年2期)2017-05-17
河南畜牧兽医(2016年24期)2016-11-29
中国资源综合利用(2016年6期)2016-01-22
兽医导刊(2015年7期)2016-01-04
云南畜牧兽医(2014年4期)2014-02-28
