
原子尺度断裂模拟进展1)
2024-03-01 08:31陈玉丽李晓雁
力学学报 2024年2期
丁 彬 高 源 陈玉丽 李晓雁
* (北京航空航天大学固体力学研究所,北京 100191)
† (清华大学工程力学系,北京 100084)
引言
断裂失效是材料在静、动态加载作用下最常见的破坏模式之一.实际应用中,厘清材料的断裂行为对材料和结构的可靠性和安全性有十分重要的意义.
材料的断裂失效行为本质在于裂纹尖端附近高应力区的存在,在连续介质力学框架下该高应力区可以由奇异的裂尖应力场来描述.若从多尺度视角看断裂,宏观上的尖锐裂纹在原子尺度存在一个最小的半径,约为原子间的化学键键长.基于该力学本质,断裂是一个多尺度的问题和过程,绝大多数断裂过程最终都涉及原子键的断裂,因此原子尺度的演化对材料的宏观断裂行为有重要影响.1921 年,Griffith[1]从能量的角度提出了断裂力学的划时代理论,认为裂纹的临界驱动力为产生新的裂纹表面所需的表面能,而表面能的概念也可以在原子尺度进行很好的理解,是由裂纹表面原子与内部原子的受力不同造成的.另外,从研究对象来看,过去的几十年里,由于制造和加工技术的快速进步,涌现了许多新型的纳米结构材料(如纳米单晶、纳米孪晶等晶体结构、金属玻璃、锂化硅电极等非晶结构、非晶硅/锂化硅界面等异质界面结构).与特征尺寸在几十微米以上的传统晶体材料相比,纳米结构材料由于其独特的微观结构,往往可以获得非凡的力学性能,例如更高的强度、更好的延展性、更高的断裂韧性和更显著的抗腐蚀、抗磨损性能.因此,断裂力学的本质和研究对象的不断更新与拓展促使我们需要从原子尺度来研究新型纳米结构材料的断裂行为和断裂机理.
近年来,针对这些新材料断裂行为的实验研究取得了重要进展[2-9].然而,由于实验和可视化技术的种种限制,仅通过实验来确定材料在最小特征尺度(通常是纳米尺度)下的变形过程或机制是十分困难的,独特的实验现象的揭示只能通过原子尺度模拟.随着计算能力的日渐强大,原子尺度模拟技术愈渐成熟,已经成为了研究纳米结构材料变形和失效的有力工具之一[10-12].由于其固有的原子尺度分辨率,原子模拟经常被用于解释实验,揭示纳米结构材料的变形行为过程和断裂机制.需要注意的是,原子尺度模拟通常选用经验势函数,但裂尖的原子结构严重畸变偏离平衡态,因此一些经验势函数并不能准确描述断裂行为,需要采用基于量子力学计算直接构建的更精确的势函数.另外,原子尺度模拟和实验在样本尺寸和加载速率方面存在着巨大差异,在解释模拟结果时需要较为谨慎.原子尺度模拟所揭示的现象和规律,例如裂尖的变形行为[13]、裂纹扩展的韧脆转变现象[14]、材料构型在循环过程中的稳定性[15-16]、断裂强度随影响因素的变化规律[17]等等,能够和实验吻合得很好,这是因为力学行为和内在变形机理在很大的应变率跨度范围内都保持一致.近年来,超级计算机和高效算法的不断发展(多尺度建模、机器学习方法等),在一定程度上缩小了这种差距.除此之外,原子尺度模拟揭示的变形和破坏机制可以通过使用扫描电子显微镜(scanning electron microscope,SEM)/透射电子显微镜(transmission electron microscope,TEM)在相同的空间尺度上进行验证[18-19].目前,原子尺度模拟被广泛应用于探索多种纳米结构材料的变形、断裂行为以及相关机制[14,20-26].原子尺度模拟不仅为微观组织演变和各种缺陷之间的相互作用提供了新的力学见解,也为新材料的工艺-结构-性能关系链提供了重要的指导.原子尺度模拟的结果和见解对设计和制造具备优异力学性能的新材料具有重要意义.
在本综述文章中,首先介绍用于原子尺度断裂的模拟计算方法,主要为原子尺度断裂模拟中常用的加载方式,以及量化抵抗断裂程度的方法,包括计算断裂韧性的几种方式.然后重点介绍了近十几年的研究进展,包括利用原子尺度模拟来研究纳米单晶、多晶和孪晶等晶体材料、金属玻璃和锂化硅等非晶材料、非晶硅/锂化硅界面等界面材料的裂纹扩展行为和断裂机制.这些原子尺度模拟提供了断裂过程中裂纹尖端的原子尺度构型的动态演化过程,揭示了断裂失效机制,如裂纹尖端的损伤形式,裂纹与各种缺陷之间的相互作用,脆性和韧性断裂行为转变的力学机理,应力错配驱动的界面自发分层原理等.这一方面有助于理解纳米结构材料独特的断裂行为,另一方面也可用于指导新纳米结构材料的制备与合成,此外对未来的研究思路提供一些思考和见解.
1 方法
对材料断裂的研究主要集中在外加载荷作用下的裂纹扩展行为.因此,进行断裂模拟的一般步骤为,首先在模拟样品中预制裂纹,然后施加适当的载荷和边界条件.随着加载过程的进行,输出与裂纹扩展有关的力学信息,如原子应力和应变,裂纹尖端位置和外加应力强度因子等,以此来分析量化裂纹扩展、微观结构和外加载荷之间的关系.
原子尺度模拟的手段主要可分为: 第一性原理计算(first principle calculations)、分子静力学(molecular statics,MS)计算和分子动力学(molecular dynamics,MD)计算.第一性原理计算是一种以电子密度分布为基本变量,研究多粒子体系基态性质的量子力学计算方法.基于密度泛函理论(density functional theory,DFT)将多体基态的解近似为基态电子密度分布的解.该方法可以较为精确地求解体系中的能量和电子结构等物理性质,但是所需要的计算资源较大,通常模拟的体系尺寸较小,时长有限.MS 仿真过程中,系统基于能量最小化算法(如共轭梯度法或最速下降法)进行弛豫,因此可以考虑系统的准静态过程,但不能考虑有限温度的影响.相比之下,使用MD 方法进行模拟期间,系统通过在给定时间步长内求解牛顿运动方程,松弛过程中考虑原子的实际运动,使得MD 模拟可以在有限温度下研究系统动力学和热力学的响应.
虽然MD 模拟相较于第一性原理计算可模拟更长的时间,但是总模拟时间仍然被限制在了~ 10 ns之内,导致加载过程中需要施加极高的应变率,通常比实验中使用的应变率高7~ 11 个数量级.幸运的是,如此高的应变率一般情况下不会对材料的潜在变形行为和破坏机制造成重大影响.在第2 章中,我们介绍了最近的一些关于纳米结构材料的裂纹扩展行为和断裂机制研究,并与相关的实验结果进行了对比,证明了原子尺度模拟的可靠性和准确性.
1.1 加载方法
在断裂模拟中,我们主要关注裂纹扩展行为和扩展引起的失效和损伤,首先需要在模拟样品的初始构型中预制裂纹.通常来说,有两种方法可以生成裂纹.第1 种方法是移除楔子形状内的原子来产生裂纹或凹槽,该方法引入的裂纹会有一定宽度;第2 种方法是解除裂纹表面两侧原子间的相互作用来产生裂纹,该方法产生的裂纹为原子尺度的尖锐裂纹.初始裂纹一般位于模拟样品的中心或边缘,对应于中心裂纹或边缘裂纹的构型.
在断裂模拟的过程中,对初始裂纹施加载荷是整个模拟的关键步骤.在实际的断裂工况中,I 型加载是主要的受载模式.因此,大多数仿真通过施加垂直于裂纹面的拉伸载荷,来模拟I 型裂纹的扩展.本文主要介绍MD 模拟中4 种I 型裂纹加载的具体方法(见表1).这些方法对应于实验中的I 型裂纹加载方式,因此对于脆性材料和韧性材料在理论上均适用,但需要根据具体情况选择合适的加载方案.为了方便介绍,下文以裂纹面位于xOz平面,加载方向为y方向为例.

表1 加载方法汇总Table 1 Summary of loading methodologies
1.1.1 均匀加载
均匀加载是通过在动态弛豫过程中更改MD 模拟盒子的体积或形状进行的.
对于I 型裂纹加载形式,首先需要在加载方向(即垂直于裂纹面的方向)施加周期性边界条件,然后以一定的应变率均匀增加模拟盒子在该方向的尺寸,以施加均匀的拉伸应变.
该方法会按照指定应变流程在每一个时间步内先进行模拟盒尺寸的拉伸,然后将拉伸位移平均到每一个原子上,并且在该时间步内系统自动弛豫.该方法使用周期性边界条件可以在一定程度上减小加载方向的边界效应.
1.1.2 速度梯度加载
速度梯度加载是通过在动态弛豫过程中以一定的速度移动边界层原子,并对模型内部施加速度梯度进行的.
在预制裂纹的模拟样品中进行I 型加载时,首先需要平行于裂纹面在样品两侧边界选取一定宽度的原子层,将原子层固定为刚体.加载时存在两种方案,第1 种为一侧原子层固定,另一侧原子层以给定的速度均匀移动;第2 种为两侧原子层以等值的速度向相反的方向均匀移动,两种方法等价.为了避免两侧原子层移动时产生的应力波,在模拟开始时根据y方向坐标对内部所有原子施加线性速度梯度分布.在整个模拟过程中,两侧刚性原子层内的原子受力为0,角动量为0,以固定的速度进行移动.所有其他原子则根据局部作用力移动.
该方法设置简单,操作简便.但是与均匀加载一样可能存在动态效应.为降低动态加载的影响,可以在每次加载后进行能量最小化弛豫,直到体系的能量变化在相邻时间步小于某一定值.
1.1.3K场加载
K场加载的方法由Sinclair 等[27-28]提出,随后得到广泛应用[25,29-33].
基于线弹性断裂力学,当应力强度因子为KI时,裂纹尖端的位移场为
式中,r为到裂纹尖端的距离,θ为相对于裂纹平面的角度,G和ν分别为材料的剪切模量和泊松比,κ是Kolosov 常数,在平面应变条件下等于3 -4ν,在平面应力条件下等于(3 -ν)/(1+ν).基于式(1)可以计算出当应力强度因子变化ΔKI时,区域中任意一点的位移增量Δux和Δuy.
当对MD 模拟体系的边界施加符合该位移场的位移边界条件时,就称为K场加载(又被称为K控制法).图1 显示了在预制裂纹的模拟样品中施加K场加载的示意图,首先根据模拟样品的构型划定边界为固定区域(fixed),通常为方形或者圆形.在每个加载步骤中,先使固定区域(fixed)的所有原子按照式(1)进行移动,然后保持固定区域原子不动,同时松弛自由区(free)内部的所有原子,松弛的时间应足够长,使总能量达到收敛.如在后续相邻时间步的样品总能量变化小于某一给定值,则认为体系已经平衡,循环上述步骤.为保证迭代的精度和收敛性,通常在进行K场加载时,应力强度因子的增量ΔKI应取较小的值,如KIC/100 甚至更小,其中KIC是裂纹扩展的临界应力强度因子.

图1 K 场加载预制裂纹的模拟样品的示意图Fig.1 Schematic of mode I loading on the sample with a pre-existing crack under a K-field in the fixed region
对固定区施加的K场会成为自由区的边界条件,为了避免K场对裂纹尖端的塑性行为造成影响,需要固定区域距离裂纹尖端较远,达成小范围屈服条件,因此模拟样品的尺寸应足够大,以裂纹尖端塑性区被限制在自由区域内为准.如果模拟的样品尺寸较小,就需要在自由区和固定区之间引入缓冲区(buffer)[12],如图1 中所示.缓冲区中的原子不会独立运动,而是根据自由区中原子的作用力进行控制,可以在一定程度上避免固定区对裂纹尖端塑性行为的影响.
基于线弹性断裂力学理论分析塑性区大小为[34]
式中,ry为塑性区长度,KI为应力强度因子,σy为屈服应力,平面应力和平面应变条件下参数b分别取1 和1/3.通过该公式可以估算塑性区的尺寸,从而合理选择固定区域(fixed)、自由区域(free)以及缓冲区域(buffer)的尺寸.
1.1.4 静水应力加载
静水应力加载是通过使模拟样品在给定静水压力下平衡,后去除模型周围静水压力使裂纹自发扩展进行的.
在利用静水压力进行I 型裂纹加载模拟时,首先需要对模拟样品加热,使其达到目标温度.然后在目标温度与合适的静水压力下弛豫足够时间,使系统达到平衡.之后在样品内生成一条尖锐裂纹.最后去除静水应力后,裂纹将自发进行扩展.
该方法通过预加载的方式为裂纹扩展提供驱动力,不依赖给定应变率进行加载.可以消除MD 模拟中常见的加载速率依赖效应.并且该方法下裂纹扩展的过程是一个稳态过程,裂纹尖端的扩展速度与实验测量的速度相似.但是,该方法需要预先试验不同的静水应力,才能得到可以使裂纹扩展而不会不稳定断裂的加载条件.
1.2 断裂能计算方法
原子尺度的断裂模拟不仅可以提供断裂过程中的力学细节,还可以用来估算材料的断裂韧性.接下来将介绍5 种基于原子尺度模拟结果计算断裂韧性的方法,包括能量释放率法、线下面积积分法、临界应力强度因子法、原子尺度内聚力模型法和原子尺度J积分方法.
1.2.1 能量释放率法
断裂能量准则认为,当裂纹扩展所需能量超过材料抗力时,裂纹即向前扩展,其中材料抗力包括表面能、塑性耗散、热耗散以及其他与裂纹扩展相关的能量耗散.断裂能量准则最初由Griffith[1]提出,后续lrwin[35]提出了更方便的应用于工程实际中的方法,即能量释放率法.
能量释放率G定义为每增加单位裂纹的表面积所减小的总势能[36-37],可以通过比较初始和最终的断裂构型进行如下计算获得
式中,ΔU是应变能的变化,ΔW是裂纹扩展有关的外界做功,Δa是裂纹扩展的长度,t是模拟试件的厚度.ΔU可以根据初始构型和最终构型的应力和应变状态来估计,ΔW可以通过对模拟过程中得到的力-位移曲线进行积分获得.当G=GC时,材料发生断裂,GC即为临界能量释放率.值得注意的是,能量释放率法在线弹性断裂力学假设下推导得出,因此较为适用于脆性材料和小变形情况下的韧性材料.
目前已有研究将该方法应用于纳米尺度石墨烯[38-39]和金属玻璃[40-41]中能量释放率的计算,得到结果与实验测量结果吻合程度较好.
1.2.2 线下面积积分法
线下面积积分法通过对加载过程中应力-应变曲线进行积分,估算断裂过程中的能量耗散,较为适用于脆性材料和韧性材料,其具体计算方法如图2所示.对完整样品和存在裂纹的样品分别进行加载,完整样品用于记录加载过程的应力-应变曲线,存在裂纹样品用于确定裂纹开始扩展时的临界应变εc,断裂能的估算值为完整样品直到临界应变时所做的功(即应力-应变曲线所围的面积),表达式如下[40,42]
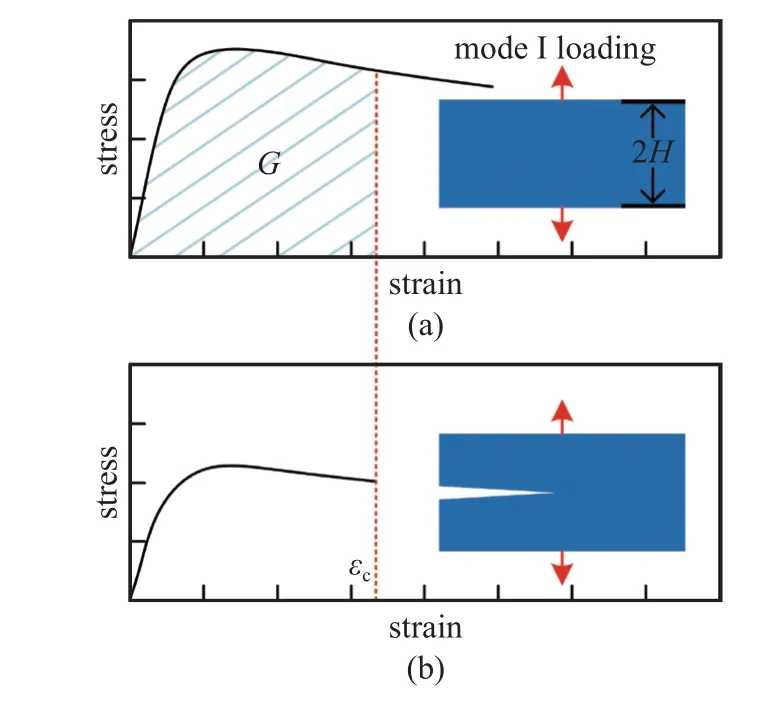
图2 线下面积积分法示意图Fig.2 Schematic of stress-strain curve integral method
式中,H是条带的半宽,εc是裂纹开始扩展时的临界应变.
对于脆性材料,式(4)可以简化为线弹性应力-应变关系
式中,E是材料的杨氏模量.
1.2.3 临界应力强度因子法
应力强度因子K是与构件几何尺寸、裂纹尺寸和外加载荷相关的函数,它表征了裂纹尖端受载和变形的强度,是判断含裂纹模型的断裂和计算裂纹扩展速率的重要参数.
对于无限大板中含中心裂纹受I 型加载的情况下,应力强度因子的计算公式如下
式中,σ 为无穷远处的均匀作用力,a为裂纹长度的一半.当K=KIC时,材料发生断裂,KIC即为临界应力强度因子.该方法基于线弹性断裂力学理论,较为适用于脆性材料和小变形情况下的韧性材料.
在原子尺度断裂模拟中,如果通过K场加载的方式施加载荷,则可以直观观察裂纹扩展过程与应力强度因子的关系,KIC即为裂纹萌生时的应力强度因子.
该方法已被用于非晶态锂化硅的原子尺度断裂模拟中[25],在对a-Li0.5Si,a-LiSi 和a-Li1.5Si 断裂韧性的分析中与实验结果吻合较好.
1.2.4 原子尺度内聚力模型
在连续介质力学中,内聚力模型(cohesive zone model,CZM)用于估算断裂界面的牵引力-张开位移关系,并且经常与有限单元法(finite element method,FEM)结合用来研究材料内部或者界面上的裂纹扩展.而在微纳尺度,结合MD 与CZM,Yamakov等[43]发展了原子尺度的内聚力模型.
该方法首先在裂纹面两侧的区域划分等体积的内聚力单元,每个单元的长度和宽度保持一致,通常约为1 nm.然后计算每一时刻每一单元对应的内聚力和位移,内聚力为单元内所有原子的应力进行统计平均,位移λ可以通过回转半径Rg来计算
式中,W为模拟试样的宽度.Rg可以通过原子坐标的计算来确定
式中,yi是原子i的y坐标,yC是单元的质心坐标.式(8)计算得出Rg代入式(7)中,λ 的正根对应于单元的张开位移.
统计裂纹起裂到试样裂纹完成断裂时刻内所有单元的内聚力以及对应的位移,将所有统计数据进行光滑拟合,得到牵引力-张开位移曲线.最后积分曲线下所覆盖的面积为界面断裂能
这种方法多用于研究界面裂纹,对于有较明显界面的脆性和韧性材料较为适用.目前已经被用来分析金属铝中沿大角度晶界界面的原子尺度脆性和韧性断裂现象[43],与相关的连续体内聚区建模结果吻合程度良好.另外该方法还被用来计算锂化硅和非晶硅之间的界面断裂能[44],与实验中的结果具有较好的一致性.
1.2.5 原子尺度J积分方法
弹塑性断裂力学中,Rice[45]提出了与积分路径无关的J积分方法.在线弹性情况下该方法与能量释放率方法等价.该方法避开了直接计算在裂纹尖端附近的弹塑性奇异应力、应变场,而用J积分作为表示裂纹尖端应力集中程度的特征参量,因此较为适用于脆性材料和韧性材料.
传统的J积分方法适用于连续介质材料中,Zimmerman 等[46-48]通过加权平均的方式,对原子尺度的离散数据进行了处理,提出了原子尺度J积分方法.首先,Eshelby 能量动量张量可表示为
式中,ϕ 为自由能密度,F为变形梯度张量,P为第一Peach-Koehler 应力.变形梯度F=∇Xχ其中当前构型x=χ(X,t).J积分直接定义为Eshelby 张量S的面域积分
代表将Eshelby 应力在法向N的分量沿域Ω的表面∂Ω进行面域积分.N为域Ω的表面∂Ω的外法线方向.对于温度恒定且处于平衡态的系统,上述表达式可以简化为
式中,H=F-I为位移梯度张量,因此第1 个等式成立要求满足应力平衡.第2 个等式要求满足没有热流动,此时自由能密度 ϕ 等效为内能密度w.
为了求得原子尺度J积分计算所对应的参量,基于Hardy 映射[49],引入连续的核函数(也称为局域化函数)ψ(X),需要满足以下两个条件
在下述说明中,希腊字母代表系统中某一特定的原子,例如,Xα表示原子α的位置.模拟体系中的能量密度可定义为每个原子能量密度 ϕα的局域加权平均
式中,ϕα为原子α的能量密度,X为原子的坐标.减去初始构型中的原子的能量密度是为了消除初始构型能量密度不均匀的影响.
模拟体系中的位移梯度可采用质量加权平均的方法进行计算
式中,uα和mα分别为原子α的位移和质量.
为了确保和连续介质力学之间的一致性,需要引入一个“连接函数”Bαβ,其表达式为
模拟体系中的第一Peach-Koehler 应力通过对原子应力进行基于核函数的加权平均来表示
式中,fαβ为原子α和β之间的相互作用力.
Zimmerman 等[46]讨论了发展的原子尺度J积分与连续介质J积分3 个方面的一致性: (1)有限元网格和原子尺度核函数尺寸的收敛一致性;(2)应变能、应力场和变形场之间的一致性;(3)评估了相关应力场进行面域积分的固有误差.并将该方法应用于K场加载下含预制裂纹的金属金的原子尺度断裂模拟仿真,所得到的结果在应力强度因子较小时,J预测值与成线性关系,与线弹性断裂力学结果吻合程度较好.
后续不断有研究将原子尺度J积分方法应用于多种材料的原子尺度断裂模拟中,如非晶锂化硅[25]、Cantor 合金[50]、石墨烯和非晶碳[33,51]等,均与文献中已有的数据吻合程度较好,该方法得到了广泛地验证.
2 典型结果
近年来,国内外学者针对众多纳米结构材料的原子尺度断裂行为和机制进行了较为深入地研究.纳米结构材料根据组分和排布主要分为晶体、非晶和界面3 类,接下来我们选取了这3 类纳米结构中的典型代表材料,介绍其通过原子尺度断裂模拟揭示的裂纹扩展行为和断裂机制等相关结果.如无特殊说明,以下结果均通过开源软件LAMMPS[10](large-scale atomic/molecular massively parallel simulator)计算得到.
2.1 晶体材料
晶体是指由大量微观物质单位(原子、离子和分子等)按一定规律有序排列的结构,具有长程有序性、均匀性、各向异性和对称性等特点,学者们对其进行了许多实验、仿真和理论上的研究.在本节中我们将介绍单晶、多晶、孪晶3 类典型的晶体材料中原子尺度断裂模拟的最新进展,主要分析了裂纹与各种缺陷(如位错和孪晶界)之间的复杂相互作用.
2.1.1 单晶材料
单晶材料指内部原子按照同一种排布方式规则排列,其晶体结构呈现连续周期性排布规律,通常强度较高,但是目前仍然存在较难制备和抵抗断裂破坏的能力较弱等缺点.接下来将以单晶硅和单晶金为例,介绍单晶材料原子尺度断裂机制方面的研究.
目前的太阳能光伏电池中,有95%由单晶硅晶片制成.但是这些电池在弯曲应力下很容易发生断裂和破坏,限制了太阳能电池的应用场景.因此开发柔性可弯曲单晶硅片具有广阔的应用前景.
最近Liu 等[52]的研究表明,织构的单晶硅片总是在边缘区域粗糙表面的表面棱锥体之间的尖锐通道处断裂,且断裂的裂纹几乎平直,如图3(a)所示.若是将该尖锐通道进行钝化处理,可以显著提高硅片的断裂临界应力,并且改变裂纹的扩展路径,并在裂纹扩展前,通过多个解理位置消耗更多的能量,如图3(b)所示.此方法可以显著提高单晶硅芯片的抗弯能力,由此制成的单晶硅电池可以完成360°的折叠而不损坏,并可在1000 次侧向弯曲循环后仍保持100%的能量循环效率.
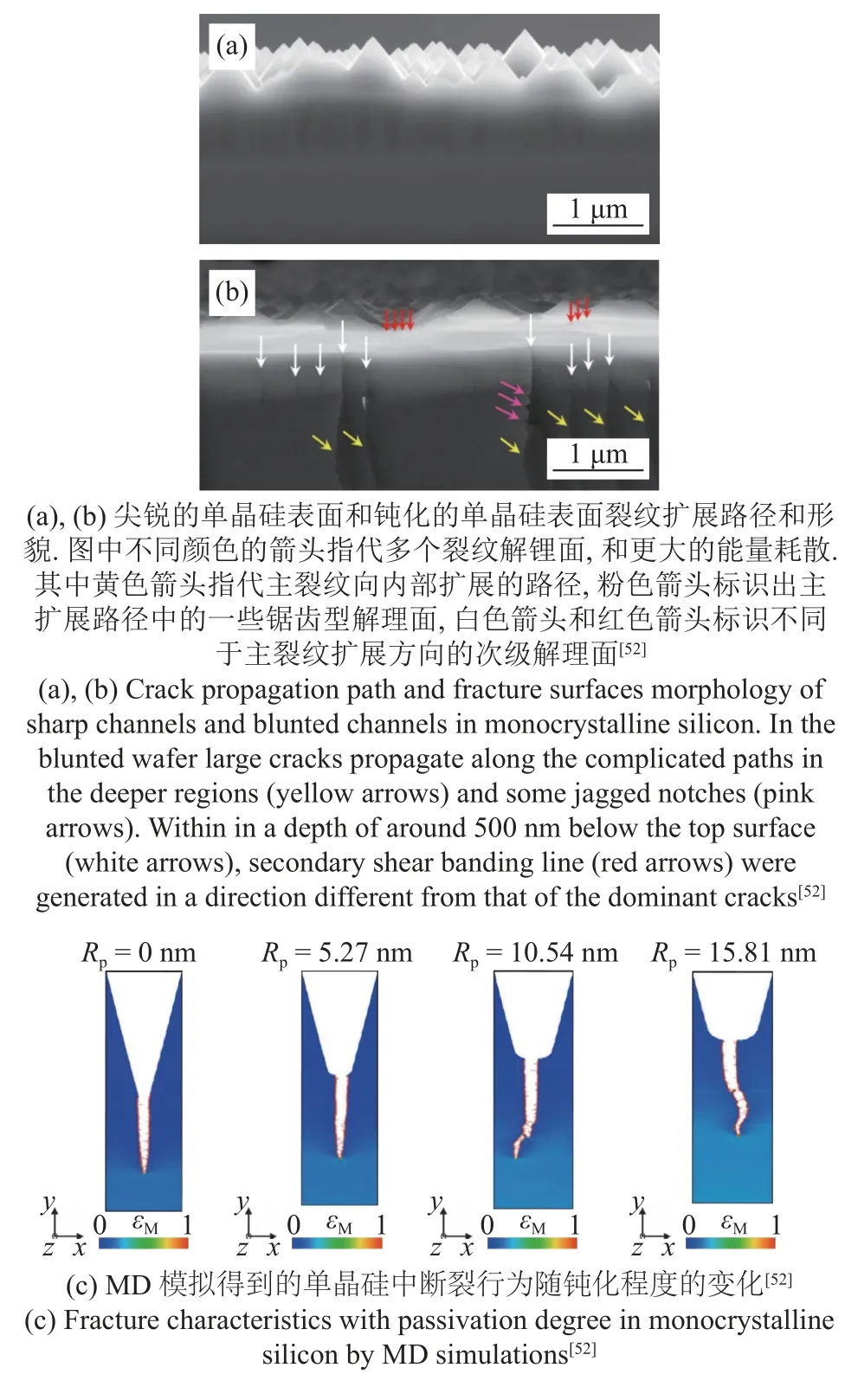
为了理解实验中发现的增韧现象并揭示背后的微观机理,文献[52]对表面棱锥体之间具有尖锐通道和钝化通道的晶体硅片进行了I 型加载的原子尺度模拟.加载方式为均匀加载,应变率为5×108s-1,势函数为Tersoff 势,该势函数可以准确描述半导体、金属和碳等材料的原子间相互作用.棱锥体间通道的曲率半径Rp由0 nm 增加到15.81 nm.模拟结果表明,具有尖锐裂纹的试件(Rp为0 nm)在9.3%的加载应变下就出现了开裂现象,而经过钝化处理的试件在17.3%的较高加载应变下才发生开裂.并且,经过钝化处理后,裂纹扩展的路径出现弯曲,对应了更高的能量耗散,如图3(c)所示.文献[52]通过原子尺度模拟证实了实验中对通道的钝化处理可以显著提升起裂的临界应变以及扩展的能量耗散,原子尺度模拟结果与实验结果高度吻合.
除了对单晶材料的断裂行为进行研究,还进行了断裂能方面的探索.为了将断裂能的计算扩展至原子尺度弹塑性材料,Zimmerman 等[46]提出了原子尺度J积分方法 (该方法在1.2.5 节中有相关介绍),并对面心立方晶胞的单晶金进行了含尖锐裂纹的I 型加载仿真,加载方式为K场加载,势函数为LJ 势(Lennard-Jones potential),该势函数由于其解析形式简单而被广泛使用.结果显示原子尺度模拟的结果与线弹性断裂力学得出的结果一致,如图3(d)所示.进一步分析表明,原子尺度J积分方法具有良好的路径无关性,如图3(e)所示,在应变较小时符合线弹性断裂力学理论,J预测值与KI2成线性关系,与线弹性断裂力学结果吻合程度较好,如图3(f)所示.
以上结果均说明,即使在应变率和样品尺寸与实验差距较大的情况下,原子尺度模拟仍然能准确地揭示材料的变形行为,并能反映出内在变形机理,是研究过程中的有力工具.
2.1.2 多晶材料
多晶材料相比于单晶材料,具有低价和产能上的优势.但是多晶材料中晶面取向不同,晶界繁杂,位错密布,晶格较多,在实际应用中带来了较多的困扰.由于深入了解晶界与缺陷对多晶材料力学性能的影响在实验上需要极高的分辨率,原子尺度的模拟方法成为了学者们有力的研究手段[53-56].这里举例介绍几项多晶材料研究的工作.
与机械剥离手段制备的石墨烯相比,化学气相沉淀(chemical vapor deposition,CVD)是一种更有前途的大规模生产技术,但该方式生产的石墨烯是多晶的,会产生沿晶界分布的五边形-七边形缺陷和相邻晶粒间的不同手性现象[57].这些缺陷可能会导致应力集中,引起裂纹扩展,削弱材料的强度[58-59].但同时,晶界的存在也可能会影响裂纹的运动,提高材料的断裂韧性.为了探究多晶石墨烯的断裂机制,Jung 等[38]对其进行了原子尺度模拟.该研究使用的势函数为AIREBO (adaptive intermolecular reactive empirical bond order)势,加载方式选用速度梯度加载,应变率为1.0×108s-1.
模拟样品考虑了晶粒的不同规则程度以及晶粒的不同尺寸.图4(a) 展示了规则颗粒(regular grain,RegG)和不规则颗粒(ir-regular grain,IrreG)组成的多晶石墨烯.在对具有相同裂纹长度的不同样品进行断裂模拟时发现,多晶石墨烯中的应力分布不像单晶石墨烯那么集中,如图4(b)所示.研究表明,裂纹尖端附近的多个晶界有助于应力的分布,使材料的变形分布较广,不集中于裂纹尖端.同时,多晶石墨烯中复杂的应力分布也使得裂纹难以快速扩展,许多裂纹在扩展开始后被阻止,如图4(c)中应力-应变曲线中的转折所示,因此材料抵抗断裂的能力较强.在对规则晶粒与无规则晶粒石墨烯进行断裂能计算时,选用能量释放率法作为断裂韧性的表征,结果发现,多晶石墨烯,尤其是无规则晶粒组成的多晶石墨烯,具有比单晶石墨烯更高的能量释放率,且能量释放率随着晶粒的尺寸减小而增加,如图4(d)所示.该工作解释了多晶石墨烯中的增韧原理,并且分析了断裂韧性与平均晶粒尺寸的关系,为气相沉淀方式制备的石墨烯提供了一个估算断裂韧性的经验法则.
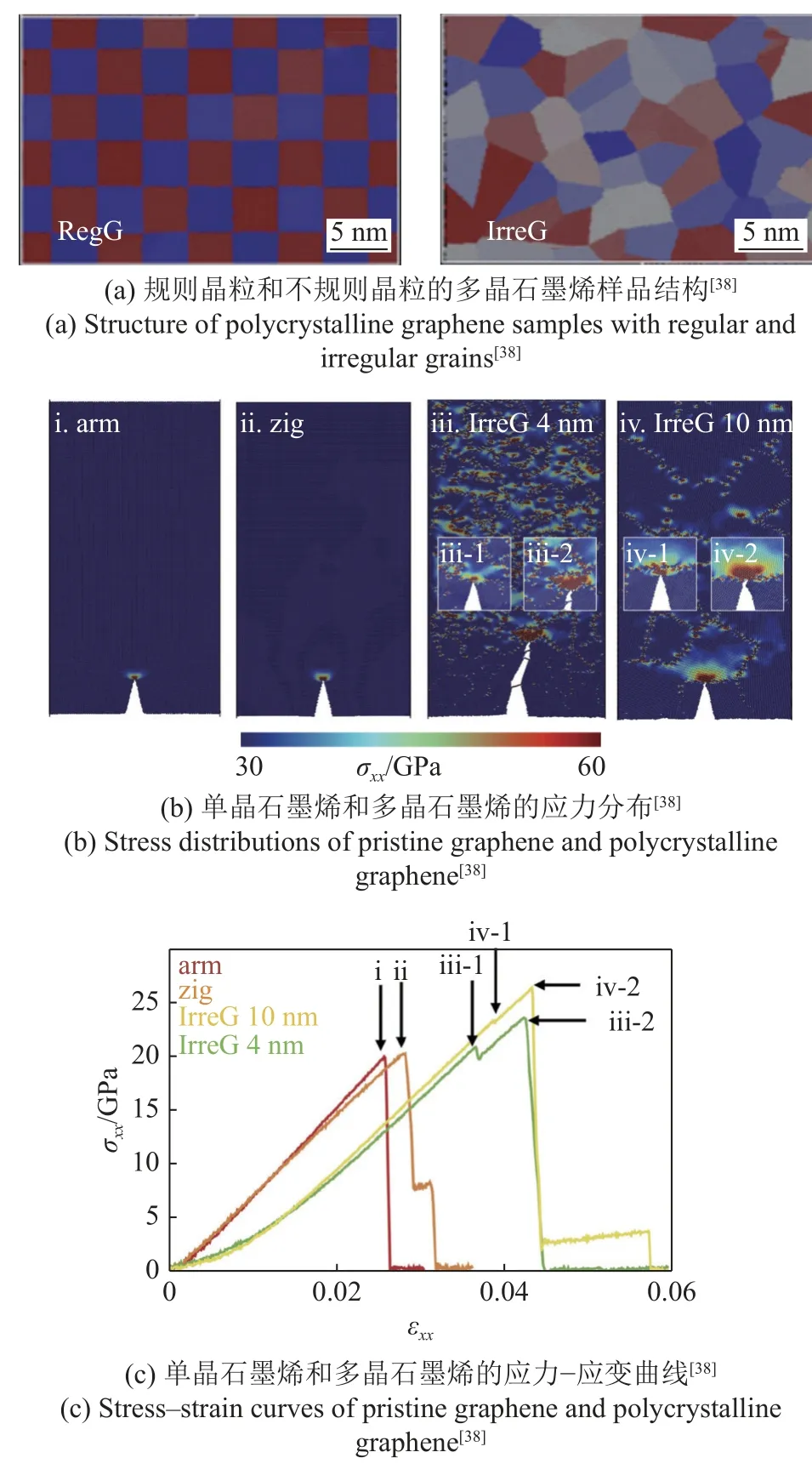
图4 多晶材料相关原子尺度断裂模拟研究Fig.4 Simulation study on related atomic scale fracture of polycrystalline materials
除二维材料外,Rudd 等[54]针对多晶铜块体材料中的裂纹扩展进行了原子尺度模拟,发现随着加载的进行,会在晶界处形核孔洞,孔洞生长优先沿晶界进行,随后与主裂纹合并导致裂纹的快速扩展,发生脆性断裂.
2.1.3 孪晶材料
孪晶材料具有层次化的微结构,在微米或亚微米大小的晶粒中嵌入高密度的纳米孪晶,可以在一定程度上提升强度与断裂韧性.
在金刚石中引入纳米孪晶微结构已被证明是提高金刚石硬度和韧性的有效方法[60-63].人们发现含有多种不同结构金刚石的纳米孪晶金刚石复合材料,表现出了极高硬度和韧性.TEM 观察表明,断裂韧性的提高与纳米孪晶、连接在一起的不同构型金刚石以及裂纹表面附近的局部相变有关.为了解释纳米孪晶金刚石材料的增韧机理,Zeng 等[64]采用原子尺度模拟对纳米孪晶金刚石材料进行了断裂机制研究.该研究的势函数为AIREBO,加载方式选用速度梯度加载,应变率约为2.0×108s-1.卸载过程通过在给定应变下对模拟样品施加相反的载荷进行.
在对单晶金刚石的模拟中,裂纹沿(111)晶面直线扩展直到完全断裂,如图5(a)所示.而在纳米孪晶金刚石中,裂纹扩展到达孪晶界时会被阻拦一段时间,如图5(b)所示,越过晶界后会出现裂纹的偏转,使得裂纹扩展路径为锯齿形路径,如图5(c)和图5(d)所示.该路径会比单晶金刚石中的直线扩展路径消耗更多的能量,从而提高材料的断裂韧性.值得一提的是,由于裂纹尖端存在较高的应力集中,可能会使得金刚石发生相变,产生无序的原子团簇,如图5(d)所示.随着裂纹的不断扩展,原子团簇将演变为原子链或原子网络,连接在两个断裂面上,充当原子链桥阻碍裂纹面的分离和裂纹的扩展,导致更多的能量耗散.当引入的纳米孪晶包含多种不同多型体的金刚石时,更易出现相变现象.

图5 孪晶材料的增韧机理和裂纹愈合现象Fig.5 Toughening mechanism and crack healing of twin materials
在对含纳米孪晶的金刚石进行卸载模拟时发现,若裂纹尖端未形成无序的原子团簇,随着卸载的进行,断口表面的碳原子重新成键连接,样品中的应力将会恢复为0,裂纹完全愈合,如图5(e)~ 图5(g)所示,该现象与实验中观察到的现象一致[63].
该研究表明,孪晶材料中的孪晶界会阻碍裂纹的扩展,且会在加载过程中引起裂纹的偏转,导致更高的能量损耗,从而提高材料的断裂韧性.为设计具有硬度-韧性协同效应的超硬材料提供了方向.
除了对穿越孪晶界裂纹的断裂模拟研究,Monismith等[65]还进行了沿孪晶界的原子尺度断裂模拟.最近有研究表明,LLZO (Li7La3Zr2O12)作为一种新型的固体电解质材料,具有较高的离子电导率[66]和稳定性[67].但是由于孪晶界存在引入的局部输运和结果不均匀性[68],会导致裂纹扩展并最终使电池出现短路.为了揭示LLZO 的晶间断裂机制,Monismith 等[65]对其进行了MD 模拟.该研究的势函数为Buckingham-Coulomb 势,加载方式为静水应力加载,断裂能计算方式使用原子尺度内聚力模型,模拟晶界为倾斜角度36.9°的孪晶界.
模拟结果如图5(h)所示,在裂纹尖端前方形核空洞,断裂方式为脆性断裂,裂纹尖端后方存在原子链桥连,增加了能量耗散.进一步研究表明,随着温度的升高,裂纹扩展机制不变,但晶格的相变过程减弱,原子链断开时锂离子的重新排序现象减弱,使得界面的断裂韧性减低.该工作将相变过程和锂离子的重新聚集看做“塑性”,作为一种无位错的耗散机制,以此解释材料断裂韧性随温度的变化.
该现象与Shin 等[69]对文石片层的研究中发现的机制类似,在含纳米孪晶的文石中,观察到当裂纹尖端到达孪晶界后,会使得周围发生相变,而在不含孪晶的文石中并未观察到这一现象.在使用线下面积积分方法计算断裂韧性后,纳米孪晶文石的断裂能比无孪晶的文石高一个数量级.这一结果与实验测量的结果一致[69].
除此之外,Kim 等[70]研究了纳米孪生铜薄膜中的裂纹扩展,发现孪晶界会充当位错墙(dislocation wall),阻碍位错的进一步扩展,并会使得裂纹沿之字形扩展,增加能量耗散.该MD 模拟的结果与实验中通过原位电子显微镜观察到的结果一致[71].
2.2 非晶材料
非晶材料是指结构无序或者短程有序而长程无序的物质,具有优异的力学、物理、化学性能.在本节中我们将介绍最近对三种典型的非晶态材料(包括金属玻璃、非晶锂化硅和非晶碳)断裂行为的原子尺度模拟,主要分析了裂纹尖端的微观结构的演化和内在变形机理.
2.2.1 金属玻璃
金属玻璃是一种新型的具有亚稳态的工程合金,通常通过快速冷却熔融合金来制备.由于非晶态原子结构,金属玻璃表现出独特的力学性能,如高弹性极限、高强度、高抗腐蚀性和抗氧化性等[72-73].
值得注意的是,金属玻璃的断裂韧性在1~80 MPa·m1/2之间剧烈波动,并表现出脆性和韧性两种断裂形式.为了揭示金属玻璃不同的断裂行为和断裂机制,Murali 等[13]对FeP (脆性)和CuZr (韧性)两种金属玻璃进行了原子尺度断裂模拟.
该研究的加载方式选用速度梯度加载,应变率为5.0×108s-1,势函数为MEAM (modified embedded atom method)势,该势函数可以准确地描述金属和合金中原子间相互作用.在对FeP 样品的模拟中,一些纳米尺度的空洞在裂纹尖端形核和聚并.随后,空洞与主裂纹合并,导致原子尺度上的尖锐裂纹快速扩展,如图6(a)所示.相反,在CuZr 样品中,裂纹前存在大范围的剪切带,使裂纹尖端钝化,如图6(b)所示.这两种不同的断裂机制分别反映了脆性断裂和韧性断裂的内在特征.
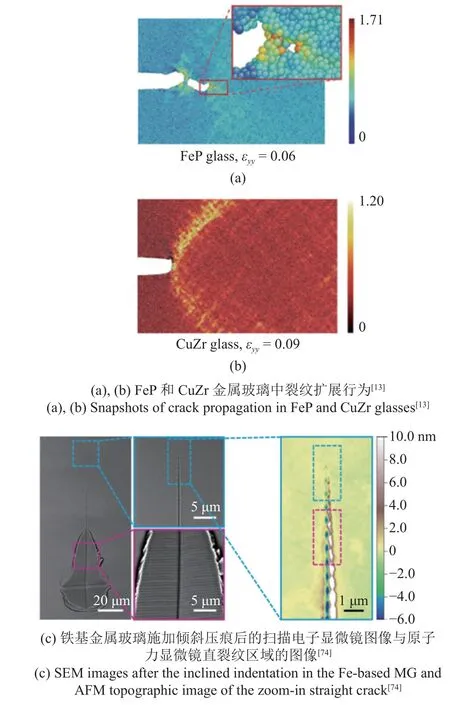
图6 金属玻璃裂纹扩展的原子尺度模拟与实验观察Fig.6 Crack propagations in metallic glasses by MD simulations and experiments
进一步分析表明,在等双轴平面应变加载下,含初始空洞的CuZr 样品,原子密度波动较小,表现为初始空洞均匀生长;而在FeP 样品中,原子密度波动较大,多个空洞在初始空洞附近成核,然后与初始空洞合并导致空洞不均匀生长.
对裂纹尖端前方的局部静水应力和发生空化的临界应力的比较分析表明,在FeP 样品中观察到的纳米级空化现象,是因为其裂纹尖端的最大局部静水应力超过了临界空化应力σc;而CuZr 样品的最大局部静水应力仍远低于临界空化应力σc,故表现为没有空洞成核的韧性断裂现象.
最近的一项实验研究中报道了针对含预制裂纹的脆性金属玻璃Fe78Si9B13样品进行纳米压痕试验[74],通过原子力显微镜(atomic force microscope,AFM)观察到了裂纹尖端的一系列纳米尺度空洞,如图6(c)所示,证明了纳米尺度空化机制理论的正确性.并且为了探索规律的普适性,研究了镁基,镧基和锆基金属玻璃的裂纹尖端形貌(断裂韧性分别为2,5,50 MPa·m1/2),均发现了不同程度的空洞形成,但锆基样品中的空洞周围存在显著的塑性流动.
为了进一步探究金属玻璃中的断裂模式的转变机理,Tang 等[75]使用修正的二元LJ (modified binary Lennard-Jones)势[40]对二元金属玻璃进行了MD 模拟,该势函数表达式如下
式中,εij是势阱深度,代表原子i和j之间相互吸引的强弱程度;σ是两原子之间的平衡距离;r是原子之间的距离.第2 项在势阱之外,即rijm<r<rijc时,添加了排斥相互作用.其中,εαβ是原子α和β之间的键能,εB是调节系数,该研究选取了0.1,0.15,0.2,0.3,0.4.εB越大,代表系统的不稳定性和脆性越大.模拟样品为矩形薄板样品,在两侧预制两个圆形凹槽,后通过速度梯度方式进行加载.研究表明在韧性较低时,样品中的断裂现象为空洞的形核和聚并导致颈缩的脆性断裂.随着样品韧性程度的增加(即εB减小),损伤主要集中在材料中的交叉剪切带上,并在样品的中心最先出现空洞的形核,并沿剪切带生长.
这些断裂模拟提供了对脆性和韧性金属玻璃失效机制的解释.
2.2.2 非晶锂化硅
由于较高的理论充电容量,硅被认为是一种很有前途的锂离子电池负极材料.在电池循环充放电过程中,大量的锂离子嵌入脱嵌到硅负极中,晶体硅逐渐转化为非晶态锂化硅.另一方面,较高的理论容量使得硅负极在循环过程中存在较大的体积变化,容易出现电极的断裂,导致电池容量的快速衰减.研究锂化硅的断裂行为和内在的断裂机理对大容量电池的设计和开发具有十分重要的意义.
最近的实验研究表明,随着锂浓度的增加,锂化硅发生了从脆性到韧性的转变[76],这表明非晶态锂化硅的断裂韧性与锂离子浓度密切相关.为了解释实验中观察到的韧脆转变的现象,我们[14]在先前的工作中进行了一系列大规模的MD 模拟,对x从0~4.4 的a-LixSi 中的裂纹扩展行为进行了研究.采用MEAM 势函数描述原子间的相互作用,加载方式为速度梯度加载,应变率为5.0×108s-1.随着加载的进行,在锂浓度较低的样品中(x≤1.71),纳米级的空洞在裂纹尖端形核、生长然后与主裂纹结合,导致原子尺度尖锐的裂纹快速向前扩展,与2.2.1 小结中讨论的金属玻璃的脆性断裂现象一致.而当锂化浓度相对较高时(x≥3.25),裂纹尖端发射剪切带,裂纹尖端变得钝化,与韧性金属玻璃的断裂现象一致.图7(a)~ 图7(d)显示了非晶锂硅合金的脆性、韧性两种断裂模式下的裂纹扩展行为.

图7 锂化硅中的脆性-韧性转变Fig.7 Brittle-to-ductile fracture transition in lithiated silicon
该研究的模拟结果与使用反应力场 (reactive force field,ReaxFF) 对含预制裂纹的a-Li0.5Si 和a-Li2.5Si 样品进行I 型加载的MD 模拟[76]中得到的结果是一致的.另外,Khosrownejad 等[25]使用K场加载的方式研究了x=0.5,1.0,1.5 的a-LixSi 样品中边缘裂纹断裂现象.在裂纹扩展过程中,这些样品裂纹尖端出现了空洞的形核和聚并,该结果与我们的模拟结果[14]相吻合.值得一提的是,该研究通过裂纹扩展时的临界应力强度因子KIC来估算断裂韧性,结果表明非晶态锂化硅的断裂韧性不仅取决于锂离子的浓度,还取决于脱锂的历史.
为了更好地解释不同锂化浓度的非晶态锂化硅中断裂机制的转变,基于弹塑性断裂力学理论,我们建立了裂纹尖端空化的判据[14]
其中σc是临界空化应力,τy是剪切屈服应力,α是与泊松比ν、拉伸屈服应变εy和硬化指数N有关的无量纲参数.为了验证上述判据,我们首先计算得到了锂化硅的临界空化应力σc和剪切屈服应力τy,并统计了裂纹尖端前方的最大静水应力和原子尺度波动导致的最小空化应力σcmin[14].图7(e)显示了随锂化浓度x的变化.其中,的值几乎恒定,与使用Prandtl 刚塑性滑移线理论计算出的1+π 的预测值接近.当x≤2 时,,而当x≥2 时,.该理论与模拟结果相一致,合理解释了断裂机制从由纳米尺度孔洞控制到由裂纹尖端发射剪切带控制的脆韧转变现象.
该工作对非晶态锂化硅的断裂机理提供了原子尺度的见解,对开发下一代稳固的电极的材料提供指导,具有深远的意义.
2.2.3 非晶碳
非晶碳相较于金刚石具有更高的断裂韧性,并且具有高硬度[77]、低摩擦系数[78]和耐磨性[79]等性质,被广泛应用于工业生产中,如电化学传感器和耐磨涂层等.但是在实际应用中,非晶碳涂层经常因剪切或弯曲裂纹而失效.因此,断裂韧性的研究对非晶碳的应用具有极其重要的意义.在实验中通过纳米压痕方法测得的断裂韧性值分布在3~ 12 MPa·m1/2之间[80-82],虽然测试样品的密度和局部结构可能会带来结果的差异,但结果之间的较大差异仍说明目前的实验手段不足以精确测量非晶碳的断裂韧性.
为了进一步研究非晶碳的断裂机制,国内外学者借助原子尺度模拟进行了一系列研究,如探究势函数对非晶碳力学行为模拟的影响[83-84],淬火速率对非晶碳断裂韧性的影响[85]等,这里主要介绍Khosrownejad 等[33]使用K场加载对密度为2.5,3.0,3.5 g/cm3的非晶碳进行原子尺度断裂模拟的研究.
该研究的势函数为Tersoff III 势的变体,该势函数可以描述键的断裂过程,加载方式为含预制裂纹的K场加载,如图8(a)~ 图8(b)所示.仿真过程中使用过阻尼形式进行准静态计算,并将温度保持在0.1 K附近,尽可能减小原子热扰动对断裂机制的影响.
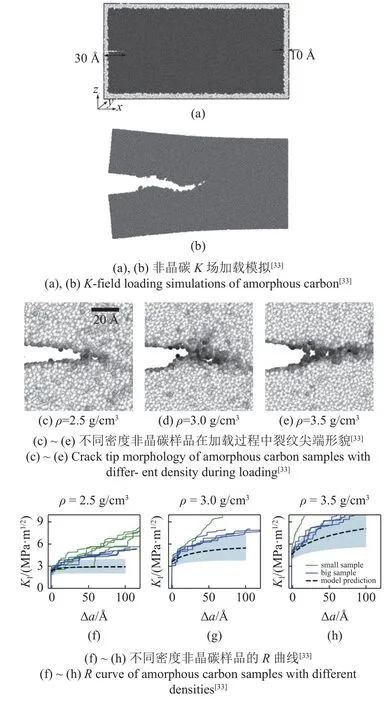
图8 非晶碳原子尺度断裂模拟研究Fig.8 Atomistic fracture simulations of amorphous carbon
仿真结果如图8(c)~ 图8(e)所示,图中使用原子配位数进行染色,阴影越深表示配位数越低,因此在颜色较深处代表有空洞的出现.可以观察到3 种密度下非晶碳的断裂机制均表现为: 在裂纹尖端前形成纳米尺度的空洞,随着加载的进行,空洞逐渐长大并与主裂纹合并,导致裂纹的快速扩展,对应于原子尺度非晶材料的脆性断裂机制.并且,空洞的尺寸和距裂纹尖端的距离一般随密度的增大而增大,但变化的幅度很小.
进一步分析K场加载的应力强度因子KI与裂纹扩展距离Δa之间的关系(即R曲线,图中蓝线和绿线分别表示大小两种尺寸的样品),得出起裂韧性在所有样品中几乎一致;R曲线斜率在两种尺寸的样品中保持一致,随着非晶碳密度的增加而增加;在接近稳态断裂韧性后,两种尺寸样品的曲线出现偏差,表现为小尺寸样品的断裂韧性更高.如图8(f)~图8(h)所示.
受限于原子尺度模拟的模型尺寸有限,在进一步加载时不满足K场加载所需的小范围屈服假设,因此无法直接通过仿真得到材料的稳态断裂韧性.Khosrownejad 等[33]通过Drugan 等[86]提出的断裂扩展准则进行合理外推,得出密度为2.5,3.0,3.5 g/cm3的非晶碳材料的断裂韧性分别为2.47~3.44,4.62~ 6.92,7.46~ 12.99 MPa·m1/2,与通过纳米压痕实验得出的3~ 12 MPa·m1/2一致[80-82].
该工作提出的对非晶碳稳态断裂韧性的分析方法,与实验数据吻合良好.并且,该方法还适用于其他具有类似性质的非晶材料,为相关领域的研究提供了思路.
2.3 界面材料
材料中存在多种多样的界面,如晶界、亚晶界、孪晶界、相界和堆垛层错等.研究界面的性质对于材料的理解和应用具有重要的意义.在本节中将介绍几项与界面相关的原子尺度断裂模拟研究.
我们[44]在先前的实验研究中发现混合Li13Si4粉末与乙醇溶液,可以一步制备大规模二维硅纳米片(Si nanosheets,SiNSs).基于实验现象猜测SiNSs的形成起源于晶态的Li13Si4粉与乙醇反应的过程中,由于锂离子迁移率的限制,Li13Si4的表面会先与乙醇反应,在微粒表面形成一层非晶态的Si,进而在未反应的Li13Si4与a-Si 之间产生一个异质界面.由于脱锂后a-Si 会倾向于体积收缩,但其变形被Li13Si4限制,从而导致了较大的错配应力,驱动了界面的自发分层,剥离出纳米尺度厚度的SiNSs.为了进一步研究Li13Si4与a-Si 界面的断裂过程,我们进行了原子尺度断裂模拟[44].该研究的势函数为ReaxFF势,加载方式为速度梯度加载,断裂能计算方式使用原子尺度内聚力模型.模拟样品如图9(a)所示,通过删除界面上的部分原子,在样品中心引入了一个椭圆形尖锐裂纹.
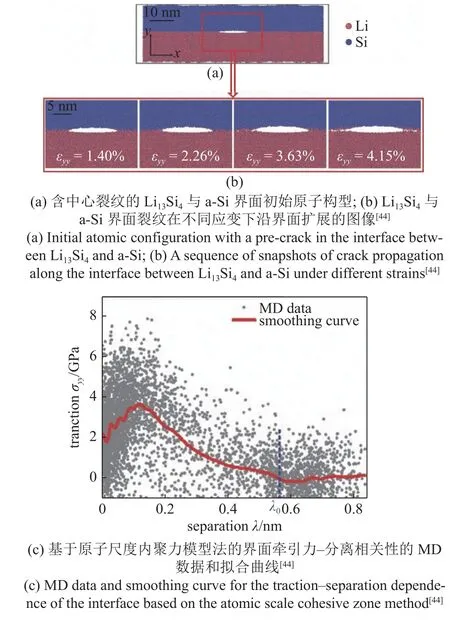
图9 界面材料的原子尺度断裂模拟Fig.9 Atomic scale fracture simulation of interfacial materials
随着加载的进行,可以观察到一些空洞在裂纹尖端前方形核,然后与主裂纹合并导致裂纹的快速扩展,如图9(b)所示.通过原子尺度内聚力模型得到牵引力-裂纹张开位移曲线,如图9(c) 所示,其中λ0为牵引力降为0 时的裂纹张开位移,表示样品完全断裂.通过对该曲线从0~λ0进行积分,估算出了界面的断裂能为1.14 J/m2.通过对半无限大模型进行分析,我们推导出硅薄膜自发分层的厚度表达式[44]
式中,Γ是薄膜-衬底体系的界面断裂能,Ef和νf分别为硅薄膜的杨氏模量和泊松比.利用该公式预测出的二维硅纳米片厚度为3~ 5 nm,与实验中的数据吻合程度较好.该工作基于实验现象建立了合理的原子尺度模型并进行了界面裂纹扩展的模拟,合理解释了纳米尺度厚度SiNSs 的形成机理,为二维纳米材料大规模合成提供了一种简便、低成本的指导方案.
除了分析化学制备领域的反应过程,许多研究使用原子尺度断裂模拟计算得到两种材料间的界面内聚力[87-90].例如,最近Wang 等[87]针对碳化硅纤维增强碳化硅(SiCf/SiC)复合材料的渐进损伤预测问题,采用MD 模拟了预制裂纹的SiC 和PyC (pyrolytic carbon)界面的界面损伤过程,据此提出了描述任意混合应力条件下界面损伤行为的CZM 模型,预测的最大外载荷与实验测得的峰值外载荷误差在6.7%以内,为损伤演化描述和本构关系预测提供了有利的工具.
3 总结与展望
本文首先介绍了原子尺度断裂模拟中常用的加载方式,其中均匀加载法和速度梯度加载法操作简便,K场加载和静水应力加载虽然操作较为复杂,但便于获取断裂阻力曲线和消除应变率效应.并总结了基于原子信息计算断裂能的方法,给出了能量释放率法、线下面积积分法、临界应力强度因子法、原子尺度内聚力模型法和原子尺度J积分法的基本原理和计算步骤.随后综述了近年来针对晶体结构、非晶结构和界面结构等典型纳米结构材料断裂行为及内在机制的研究成果.这些成果揭示了材料/结构在原子尺度的变形行为和断裂机制,并与实验中尤其是原位电子显微镜观测到的现象相吻合.此外,原子尺度模拟还可以提供一些定量的预测,如断裂韧性,其结果与实验测量值一致.
然而,尽管原子尺度模拟发展迅速,但由于在长度尺度和时间尺度上的固有局限性,以及原子间相互作用势的精度局限性,原子尺度模拟与实验之间仍然存在着明显的差距,例如原子尺度断裂模拟多采用准三维预制平直裂纹的模型,应变率比实验中使用的加载速率高出7~ 11 个数量级,这使得对于材料性能与应变率有关的材料进行分析时,原子尺度模拟将仅能提供一些定性的指导.值得庆幸的是,随着超级计算能力的不断提高和多尺度建模技术的出现,以及越来越精确的原子间势函数,这种差距在不断缩小.更强大的计算能力允许原子尺度模拟以更慢、更真实的应变率和更大的长度尺度模拟材料的变形失效过程.另外,将量子力学计算、原子尺度模拟和宏观连续介质力学模拟相结合提出的一些跨尺度方法[91-92],不仅连接了不同的空间尺度,而且全面捕捉材料从原子键断裂到裂纹扩展,再到裂尖弹性场和塑性场相互作用的复杂断裂过程.用于模拟的原子间势函数也变得越来越精确和可靠,特别是基于键级的ReaxFF 势函数[93]已经可以描述原子键的形成和断裂,该势函数比经验势函数更加准确,可以描述~ 100 nm 左右的系统.最近,随着人工智能的发展,机器学习方法[94]也被用于开发原子间势函数.该方法可以提供接近于量子力学计算的精确度,并且计算效率比DFT 计算高得多.
这些技术的快速发展无疑将使原子模拟与实验条件越来越贴合,预测值越来越准确,辅以研究人员根据物理规律的判断,将揭示出许多材料在原子尺度的变形行为和断裂机制,有助于研究下一代先进材料或结构的工艺、微结构、性能、变形和失效.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
铝加工(2020年6期)2020-12-20
中国海上油气(2019年5期)2019-10-24
科学中国人(2018年8期)2018-07-23
焊接(2016年7期)2016-02-27
断块油气田(2014年5期)2014-03-11
机械制造与自动化(2014年1期)2014-03-01
智慧与创想(2013年6期)2013-08-27
